

Fachpublikationen
| Autor/-en | Titel | Download |
|
Chloé A K Blavier, Martin H Villet, Annette Zschiesche, Volker Auwärter, Matthias Graw, Christoph Geffert, Olwen C Groth |
Entomological consequences and toxicological detection of synthetic cannabinoid receptor agonists (SCRAs) in necrophagous larvae (Diptera: Calliphoridae) Int J Legal Med. 2026 Jan 2. doi: 10.1007/s00414-025-03688-8. |
Link |
|
Detlef Thieme, Aniko Krumbholz, Martin Bidlingmaier, Christoph Geffert, Annika Hameder, Andreas Stöver, Matthias Graw, Annekathrin M. Keiler |
Influence of ethanol consumption and food intake on serum concentrations of endogenous steroids Steroids, Volume 201, January 2024, 109331. |
Link |
| K. Ruppert, C. Geffert, H.-W. Clement, C. Bachmann, M. Haberhausen, E. Schulz, C. Fleischhaker, M. Biscaldi-Schäfer |
Therapeutic drug monitoring of atomoxetine in children and adolescents with attention-deficit/ hyperactivity disorder: a naturalistic study |
Link |
| H. Clement, A. Meyer, L. Böhm, J. Böckmann, C. Geffert, C. Fleischhaker, E. Schulz | Oral fluid venlafaxine and clozapine levels for therapeutic drug monitoring Pharmacopsychiatry 2019; 52(02): 106. |
Link |
| Janina Noster*, Martin B. Koeppel*, Marie Desnos-Olivier, Maria Aigner, Oliver Bader, Karl Dichtl, Stephan Göttig, Andrea Haas, Oliver Kurzai, Arthur B. Pranada, Yvonne Stelzer, Grit Walther, Axel Hamprecht (2021) | Bloodstream infections caused by Magnusiomyces capitatus and Magnusiomyces clavatus: epidemiological, clinical and microbiological features of two emerging yeast species. Antimicrobial agents and chemotherapy, AAC-01834. | Link |
| Koeppel, M.B.; Glaser, J.; Baumgartner, T.; Spriewald, S.; Gerlach, R.G.; von Armansperg, B.; Leong, J.M.; Stecher, B. |
Scalable Reporter Assays to Analyze the Regulation of stx2 Expression in Shiga Toxin-Producing Enteropathogens. |
Link |
| Dichtl, K.; Zimmermann, J.; Koeppel, M.B.; Böhm, S.; Osterman, A. |
Evaluation of a Novel CLIA Monotest Assay for the Detection of Anti-Hepatitis E Virus-IgG and IgM: A Retrospective Comparison with a Line Blot and an ELISA. |
Link |
|
Sabrina Mühlen, Isabell Ramming, Marina C. Pils, Martin Koeppel (Labor Staber), Jana Glaser, John Leong, Antje Flieger, Bärbel Stecher, Petra Dersch |
Identification of Antibiotics That Diminish Disease in a Murine Model of Enterohemorrhagic Escherichia coli Infection | Link |
|
Karl Dichtl, Martin B. Koeppel, Claus-Peter Wallner, Thomas Marx, Johannes Wagener, Ludwig Ney |
"Food poisoning: an underestimated cause of Boerhaave syndrome" |
Link |
| Christoph Geffert: Staber Laboratory; Detlev Ranke, Henrik Winkler, Oliver Midasch: Chromsystems Instruments& Chemicals |
"Validation of a New LC-MS/MS Assay for the Analysis of Drugs in Urine and Comparison with Established Analytical Methods (GC-MS and LC-MS/MS): Advantages for the Daily Laboratory Routine" (Poster) |  |
| Dr. med. Bernd Maire, Labor Staber |
"Bisalbuminanämie - Nur eine Laune der Natur?" |
|
| Dr. med. Bernd Maire, Labor Staber; Dr. med. Kathrin Schlüter | "Problem mit dem Trenngel im Blutentnahmeröhrchen beim Multiplen Myelom" | |
| Sandra Gläser und Christoph Geffert. Labor Staber| Abteilung Toxikologie |
"Monitoring des Tabakkonsums: Evaluierung einer LC-MS/MS-Applikations-Vorschrift zur Analytik von Nicotin und Cotinin im Urin" | |
| Oliver Weimer (AHG Klinik, Wilhemsheim) und Christoph Geffert. Labor Staber| Abteilung Toxikologie |
"Hohe Ethylsulfat-Konzentration im Urin nach oraler Aufnahme eines Desinfektionsmittels" | |
| Alexander Garthof, Milos Horcicka und Christoph Geffert. Labor Staber|Abteilung Toxikologie |
"Entwicklung und Validierung einer effizienten LC-MS/MS- Methode auf Basis der MassTox Series Analytik zur Bestimmung von Amphetaminen im Urin." | |
| Dr. med. Bernd Maire Labor Staber |
"Erkrankungen der Schilddrüse Laborwerte im Zentrum der Diagnostik" (Der Allgemeinarzt 2009) |
|
| Dr. med. Bernd Maire Labor Staber |
"Fallstricke der Schilddrüsenfunktionsdiagnostik - Wechselwirkungen mit Medikamenten nicht schilddrüsenbedingter Erkrankungen und mit anderen Störfaktoren." (Abbott Times 01/2009) |
|
| Dr. med. Bernd Maire Labor Staber |
"Labordiagnostik bei Schilddrüsenerkrankungen" (Abbott Times 01/2009) |
|
| Dr. med. Bernd Maire Labor Staber |
"Doppelinfektion mit Salmonella Typhimurium und Cryptosporidium nach Türkeiurlaub" (MIKROBIOLOGE 17.Jg. 2007) | |
Fachinformationen - Endokrinologie
Adipositas und Diabetes bei Kindern und Jugendlichen
Die Zahl der Kinder und Jugendlichen mit Übergewicht nimmt in den letzten Jahrzehnten dramatisch zu. Die Folge - immer mehr Jugendliche und junge Erwachsene sind an Diabetes Ty 2 (DMT2) erkrankt. In den letzten Jahren zeigt sich, dass sich dieser Diabetestyp weltweit immer öfter schon in Jugend- und Erwachsenenalter - parallel zur Zunahme der Adipositas - manifestiert.
Prof. Dr. med. Thomas Reinehr, pädiatrischer Endokrinologe an der Vestischen Kinder- und Jugendklinik Datteln, Universität Witten/Herdecke, verweist in einem Beitrag für die Zeitschrift "MMW-Fortschritte der Medizin" auf erschreckende Zahlen: dreizehn Prozent der Jugendlichen und jungen Erwachsenen sind übergewichtig, sechs Prozent sogar adipös. Ca. 180 000 Jugendliche und junge Erwachsene müssen mit einem BMI von über 35 kg/m² als extrem adipös bezeichnet werden*.
Die Fettleibigkeit nimmt parallel zur DMT2-Häufigkeit zu, das heißt, eine bedeutende Zahl von adipösen Jugendlichen und jungen Erwachsenen können einen bislang nicht diagnostizierten DMT2 aufweisen. Da die Erkrankung weitgehend asymptomatisch verlaufen kann, wird ein gezieltes Screening in Risikogruppen benötigt.
Laut Leitlinien-Kinderdiabetologie** von 2009 gelten im Kindes- und Jugendalter folgende Risikofaktoren für DMT2:
- Typ-2-Diabetes bei Verwandten 1.-2. Grades
- Zugehörigkeit zu einer Gruppe mit erhöhtem Risiko (z.B. Ostasiaten, Afroamerikaner, Hispanier)
- extreme Adipositas (BMI >99,5 Perzentile)
- Zeichen der Insulinresistenz (arterieller Hypertonus, Dyslipidämie, erhöhte Transaminasen, Polyzystisches Ovarialsyndrom, Acanthosis nigricans)
Ein oraler Glukosetoleranztest zur Früherkennung von DMT2 soll ab dem 10. Lebensjahr bei Übergewicht BMI >90. Perzentile) und Vorliegen von mindestens zwei Risikofaktoren erfolgen.
Zur Diagnose von Typ-2-Diabetes gelten folgende Grenzwerte. Bei Überschreiten dieser Grenzwerte ist das Ergebnis bei asymptomatischen Patienten durch einen 2. Test an einem weiteren Tag zu bestätigen **:
- Nüchternglukose: >160 mg/dl (>7,0 mmol/l)
- oraler Glukosetoleranztest: 2h-Wert >200 mg/dl (> 11,1 mmol/l)
Ein HbA1c-Wert von ≥ 6,5 Prozent weist auch auf eine Diabeteserkrankung hin, es sei aber laut Prof. Reinehr unklar, inwiefern sich diese Messverfahren zum Screening eigne.
Bei neu Diagnostizierten Typ-2-Diabetes soll die Diagnostik möglicher Komorbiditäten und diabetesbedingter Komplikationen erfolgen**:
- Blutdruckmessung
- Nüchtern-Lipidprofil mit Bestimmung von Cholesterin, HDL, LDL und Triglyzeriden
- Bestimmung der Transaminasen
- Albuminausscheidung
- Augenhintergrunduntersuchung in Mydriasis
Laut Prof. Reinehr sind der Typ 1- und Typ 2 Diabetes keine sich ausschließenden Erkrankungen, es gebe Überlappungen. Auch bei übergewichtigen jungen Erwachsenen mit Typ 1-Diabetes sei eine gesteigerte Insulinresistenz zu beobachten. Hierfür stehen Bestimmungen wie Insulin, C-Peptid, intaktes Proinsulin, HOMA-Index, Insulin-Sensitivitäts-Index (Matsuda-Index) zusätzlich zur Verfügung.
Hinweise zur Abgrenzung des Typ-2 Diabetes vom Typ-1 Diabetes können folgende Laboruntersuchungen liefern**:
- C-Peptid
- diabetesspezifische Autoantikörper (GAD, IA2, ICA und IAA)
Da andere DIabetesformen in dieser Altersgruppe häufiger als DMT2 vorkommen, sollte aufgrund der Bedeutung für Therapie und Langzeitprognose die genetische Abklärung der häufigsten MODY-Formen (Maturity Onset Diabetes of the Young) bei begründetem Verdacht erfolgen**.
Die Abklärung eines MODY ist insbesondere bei Manifestationen vor dem 25. LJ ohne diabetesspezifische Autoantikörper und ohne Adipositas indiziert.
Literatur:
ÄrzteZeitung 2015 Jan 21; 6-13D; IV
* MMW Fortschr Med. 2014 Apr 30; 156 (8); 57-60
** https://www.diabetes-kinder.de/modularx/include/module/dateimanager/data/leitlinie-kinderdiabetologie-2009.pdf
CT-proAVP/Copeptin - Ein neuer Biomarker bei Verdacht auf Diabetes insipidus
Biologie
Vasopressin ist auch als ADH (Antidiuretisches Hormon) bekannt und besitzt zwei wesentliche Hauptfunktionen
- Retention von Körperwasser
- Vasokonstriktion
Es spielt bei zahlreichen Krankheitsbildern eine zentrale Rolle (u. a. Diabetes insipidus, SIADH (Syndrom der inadäquaten ADH-Sekretion), Polyurie/Polydipsie Syndrom, Hyponatriämie). Eine erhöhte Plasmaosmolalität bzw. verringertes Blutvolumen führen zu einer Ausschüttung von ADH. Die Aussagekraft von Vasopressin (ADH) als bisherige Analysemethode der Wahl war durch Bindung an Thrombozyten und die strikte Einhaltung präanalytischer Vorgaben zeitaufwändig und kompliziert.
CT- proAVP (C-Terminales Pro-Arginin-Vasopressin) ist ein aus 39 Aminosäuren bestehendes Glykopeptid, das demselben Vorläufermolekül wie Vasopressin entstammt. Es konnte gezeigt werden, dass die Konzentration von ADH und CT-proAVP direkt miteinander korrelieren.
CT-proAVP spiegelt die ADH- Sekretion wieder, bei deutlich verbessertem analytischem als auch diagnostischen Handling.
Vorteile der Bestimmung von CT-proAVP
- keine Bindung an Thrombozyten, damit keine Ergebnisverfälschung
- Messung mittels sensitivem Sandwich-Immunoassay
- Deutlich verkürzte Untersuchungsdauer
- Geringeres Probemvolumen
Klinische Aspekte/Diagnostik
Die Bestimmung von CT-pro-AVP trägt entscheidend zur Differentialdiagnostik des Polyurie/Polydipsie- Syndroms bei. Hier besteht ein wichtiger Aspekt in der Unterscheidung des zentralen vom renalen Diabetes insipidus. Bereits mit einer einmaligen Serum-Analyse von CT-proAVP gelingt die sichere Differenzierung (siehe Diagnostik-Schema). Für die weiterführende Labordiagnostik dient eine Messung des CT-proAVP in Verbindung mit Serum-Na+ nach 16h Durstversuch.
Referenzbereich außerhalb eines Durstversuches
| Osmolalität mosmol/kg | CT-proAVP pmol/l |
| 270-280 | 0,8 - 11,6 |
| 281-285 | 1,0 - 13,7 |
| 286-290 | 1,5 - 15,3 |
| 291-295 | 2,3 - 24,5 |
| 296-300 | 2,4 - 28,2 |
Diagnose-schema Durstversuch
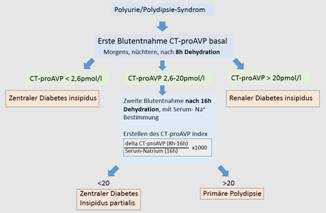
Indikation
Polyurie-Polydipsie- Syndrom
Syndrom der inadäquaten ADH- Sekretion
Verdacht auf ektope ADH- Sekretion
Anforderung
CT-proAVP (Copeptin)
Aufgrund der Korrelation des CT-proAVP mit der Serumosmolalität erfolgt die Bestimmmung der Serumosmolaslität simultan
Bestimmung von ADH entfällt ab sofort
Material
Serum
Literatur
1. Balanescu S, Kopp P, u.a. Correlation of plasma copeptin and vasopressin concentrations in hypo-, iso-, and hyperosmolar states. J Clin Endocrinol Metab. 2011 Apr;936(4):1046-52
2. Fenske W, Quinkler M, u.a. Copeptin in the differential diagnosis oft he polydipsia-polyuria Syndrom- revisiting the direct and indirect water deprivation tests, J Clin Endocrinol Metab. 2011 May;95(5):1506-15
3. Broschüre/Infomaterial „CT-proAVP“ Thermo Scientific
4. Abb. Diagnoseschema nach Thermo Scientific
Anti-Müller-Hormon: Marker der Fertilitätsdiagnostik
Physiologie
Während der Embryonalentwicklung wird die Geschlechtsdifferenzierung durch das Anti-Müller-Hormon (AMH) bestimmt. Bei männlichen Feten wird das AMH durch die Sertolizellen des Hodens gebildet. Es fördert die Rückbildung der Müller- Gänge und bewirkt somit eine normale Entwicklung der männlichen Genitalien. Bei weiblichen Feten fehlt dieses Hormon. Dies führt zur Ausbildung der weiblichen inneren Geschlechtsorgane.
Bei Frauen wird das Anti-Müller-Hormon erst mit Beginn der Pubertät von den Granulosazellen heranwachsender Follikel gebildet. Follikel im Endstadium des follikulären Wachstums, die unter direkter FSH-Regulation stehen, bilden das AMH nicht mehr, siehe Abbildung 1. Die Serumkonzentration von AMH weist eine enge Korrelation mit der Zahl der antralen Follikel auf und spiegelt die ovarielle Reserve entsprechend dem Primordialfollikel-Pool wider.
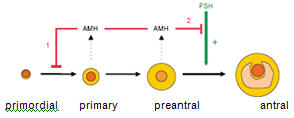
Abb. 1 Bildung und Wirkung von AMH aus Visser J et al.
Ab dem 30. Lebensjahr nimmt die Konzentration kontinuierlich bis auf in der Menopause nicht mehr messbare Werte ab. Das AMH fällt postovulatorisch nur gering ab. Der Abnahmezeitpunkt innerhalb des Zyklus ist daher ohne klinische Relevanz für die Beurteilung.
Ovarielle Funktionsreserve / Menopause
Der Eintritt der Menopause variiert stark um das 50. Lebensjahr und wird individuell durch den Verbrauch an Eizellen im Ovar determiniert. Die Bestimmung des AMH ermöglicht im Vergleich zur Bestimmung von FSH, Inhibin B und Östradiol, die erst bei bereits eingetretenen Zyklusunregelmäßigkeiten signifikante Veränderungen aufweisen, schon relativ früh eine prognostische Einschätzung der ovariellen Follikelreserve. Eine AMH-Konzentration von 0,8-1,0 ng/ml weist auf eine allmähliche Erschöpfung der ovariellen Restfunktion hin. Die Fertilität der Frau ist somit zunehmend eingeschränkt. Im Laufe der folgenden 3 Jahre tritt bei diesen Werten wahrscheinlich die Menopause ein. Die Bestimmung des AMH ermöglicht die Voraussage des Zeitpunktes der Menopause genauer als allein anhand des Alters der Frau. Dies ist insbesondere hinsichtlich der zunehmend späteren Familienplanung für Frauen ab dem 30. Lebensjahr von Interesse.
In-vitro-Fertilisation (IVF)
Im Vorfeld einer IVF ist das AMH ein nützlicher Parameter für die Vorhersage des Ansprechens einer ovariellen Stimulation. Erniedrigte Werte sprechen für eine eingeschränkte ovarielle Funktionsreserve und ein schlechtes Ansprechen auf eine Stimulation. Diese Patientinnen benötigen signifikant höhere FSH-Dosen als Frauen mit hohen Konzentrationen.
Indikationen:
- Familienplanung bei Frauen ab dem 30. Lebensjahr
- klimakterische Ovarialinsuffizienz
- Sterilitätsbehandlung
Referenzbereich
1 – 5 ng/ml: normale ovarielle Funktionsreserve
0,8 –1,0 ng/ml: ovarielle Restfunktion
< 0,4 ng/ml: Menopause
erhöhte Werte: Hinweis auf PCO (unterdrückte Follikelreifung durch erhöhte männliche Hormonkonzentration)
Material / Methode / Abrechnung
1 ml Serum, Enzym-Immunoassay
Die Untersuchung ist eine Kassenleistung
Literatur
- Visser J et al.: Anti-Müllerian hormone : a new marker for ovarian function. Reproduction, 2006, 131:1-9
- Disseldorp J et al.: Relationship of Serum Anti-Mullerian Hormone Concentration to Age of Menopause. J Clin Endocrinol Metab. 2008 Mar 11. [Epub ahead of print]
- La Marca A et al.: Anti-Müllerian hormone measurement on any day of the menstrual cycle strongly predicts ovarian response in assisted reproductive technology. Human Reproduction, 2007;22(3):766-771
Fachinformationen - Gerinnung
Thrombophilie-Diagnostik
Tiefvenenthrombosen (TVT) zählen zu den häufigen und zum Teil schwerwiegenden Erkrankungen. In der Allgemeinbevölkerung liegt die Inzidenz der symptomatischen TVT bei ca. 1/1000 pro Jahr. Ab dem 50. Lebensjahr steigt die Inzidenz exponentiell an.
Die Gerinnungsneigung (Thrombophilie) wird erhöht durch:
- Disponierende Faktoren wie Immobilisierung, Trauma, Infektion, Operation, Malignom, Kollagenose, positive Familienanamnese, Schwangerschaft oder Wochenbett
- Vorliegen von messbaren Thrombose-Risikofaktoren (s. Tabelle), die angeboren oder erworben sein können
| RIsikofaktoren | Analyse | Material | Prävalanz Gesunde % | Prävalanz Kranke % | Relatives Risiko |
|---|---|---|---|---|---|
| Verminderte APC-Resistenz * angeboren / erworben | APC-Resistenztest | Citrat-Plasma | |||
| Faktor-V-Leiden-Mutation Heterozygot Homozygot | Faktor-V-Leiden- Mutation (G1691A) | EDTA-Blut | 5 0,1 | 19 - 40 3 - 4 | 5 - 7 30 - 80 |
| FII-Mutation G20210A Heterozygot Homozygot | FII (Prothrombin)- Mutation | EDTA-Blut | 3 < 0,02 | 7 - 16 0,2 | 2 - 3 28 |
| FV/FII-Mutation kombiniert | EDTA-Blut | < 0,1 | 2 - 5 | 20 - 36 | |
| Antithrombin-Mangel angeboren / erworben | Antithrombin | Citrat-Plasma | < 0,1 | 1 - 5 | 10 - 100 |
| Protein C-Mangel angeboren / erworben | Protein C | Citrat-Plasma | < 0,5 | 3 - 5 | 3 - 20 |
| Protein S-Mangel angeboren / erworben | Protein S | Citrat-Plasma | < 0,1 | 2 - 4 | 2 – 20 |
| Persistierende Phospholipid- Antikörper | 1. Lupusantikoagulans 2. Anticardiolipin-AK 3. ß2-Glykoprotein-AK | Citrat-Plasma Serum Serum | 1 11 | 8 18 | 11 3 |
| Persistierende FVIII- Erhöhung | FVIII | Citrat-Plasma | 11 - 20 | 25 - 35 | 5 |
| Hyperhomocysteinämie | Homocystein ggf. MTHFR-Mutation | Serum EDTA-Blut | 5 - 33 | 7 - 40 | 1 - 2 |
| Disfibrinogenämie Hyperfibrinogenämie | Fibrinogen, Thrombinzeit | Citrat-Plasma |
(nach Lindhoff-Last et al. 2008, Zotz et al. 2011, Barthels (Hrsg.) 2013)
* Einer APC-Resistenz liegt meist eine Faktor-V-Leiden-Mutation (G1691A) zugrunde, selten andere Mutationen oder erworbene Ursachen.
Darüber hinaus können in besonderen Konstellationen weitere Analyte in Betracht gezogen werden, die als schwache oder kontrovers diskutierte Risikofaktoren gelten (z.B. dauerhaft und deutlich erhöhte Gerinnungsfaktoren FII (Prothrombin), FIX, FXI).
Indikationen für die Thrombophilie-Diagnostik
- Venöse Thromboembolie (VTE) vor dem 50. LJ mit ungewöhnlicher Lokalisation oder unter Antikoagulation
- Prüfung der Anlageträgerschaft
• nach VTE bei einem verwandten 1. Grades vor dem 50. Lebensjahr
• bei einem bekannten familiären Inhibitor-Mangel: Antithrombin, Protein C oder Protein S - Wunsch nach hormoneller Kontrazeption
• Bei familiärer oder eigener Thromboseanamnese
• Bei Vorliegen von dispositionellen VTE-Risikofaktoren - Schwangerschaftswunsch bei Zustand nach Thrombose
- Thrombose in Schwangerschaft oder Wochenbett
- Arterielle Thrombose vor dem 50. Lebensjahr oder bei Fehlen von Risikofaktoren
- Hautnekrose bei oraler Antikoagulation mit VKA
Relative Indikationen
- Prüfung der Anlageträgerschaft bei familiären FV-Leiden-Mutationen oder FII-Mutation
- Wunsch nach hormoneller Kontrazeption
- Therapie mit prokoagulatorischen Medikamenten bei positiver VTE-Anamnese
- Schwangerschaftsmorbidität
a. Rezidivierende Spontanaborte (2-3) vor der 12. SSW
b. Spätabort nach der 12. SSW
c. Plazentainsuffizienz, Präeklampsie, HELLP-Syndrom, intrauterine Wachstumsstörung
Zeitpunkte der Untersuchungen
| Akute VTE | 2 Mon. nach VTE | VKA | Heparin | 4 Wo. nach VKA 2 Wo. nach Heparin | Gravidität | KOK | POP | Nachweise für Diagnose | |
| Protein C | - | + | ♦ | - | + | ► | ► | ► | 3x |
| Protein S | - | + | ♦ | + | + | ♦ | ♦ | ► | 3x |
| Antithrombin (AT) | - | + | ► | - | + | - | 3x | ||
| APC Resistenz | + | + | + | - | + | + | ♦ | 1x | |
| Faktor-V-Mutation | + | + | + | + | + | + | + | + | 1x |
| Faktor-II-Mutation | + | + | + | + | + | + | + | + | 1x |
| Homocystein | + | + | + | + | + | + | + | + | 2x |
| Faktor-VIII | - | + | + | - | + | ► | ► | ♦ | 3x |
| Phospholipid-AK Anti-Cardiolipin-AK ß2-Glykoprotein-AK Lupusantikoagulans | + + - | + + + | + + - | + + - | + + + | + + + | + + + | + + + | 2x 2x 2x |
+: Untersuchung möglich
- ♦ ►: Untersuchung nein, da Ergebnis beeinflusst
VTE= venöse Thromboembolie, VKA= Vitamin-K-Antagonisten, KOK= kombinierte Pille, POP= Progesteron
Hinweis: Eine genetische Untersuchung liegt vor, wenn die Untersuchung mit der expliziten Fragestellung nach einer bestimmten genetischen Eigenschaft veranlasst wird. Genetische Untersuchungen sind mit einem speziellen Auftragsschein zu veranlassen, der die Aufklärung über
die genetische Untersuchung und die schriftliche Zustimmung des Patienten dokumentiert.
Wirksamkeit von Clopidogrel - CYP2C19 - Genotypisierung
Medikament
Clopidogrel (Plavix®, Iscover®) ist ein Thrombozytenaggregationhemmer und wird u.a. bei akutem Koronarsyndrom und bei Implantation eines koronaren Gefäßstents nach kardialem Infarkt eingesetzt. Trotz dieser Therapie kann jedoch das Risiko für ein erneutes kardiovaskuläres Ereignisses bei einem Teil der behandelten Patienten relativ hoch bleiben (sog. Non-Responder).
Pharmakogenetik
Einer der Gründe für die Clopidogrel-Resistenz ist die genetisch bedingte Variabilität im Cytochrom-P450-Enzymsystem. Clopidogrel wird zunächst als inaktive Prodrug in den Körper aufgenommen und dann durch verschiedene Cytochrom P450-Enzyme in zwei Schritten in seinen aktiven Metaboliten umgewandelt. Hierbei spielt CYP2C19 eine Hauptrolle, da es in beide Metabolisierungsschritte involviert ist.
Polymorphismen in diesem Gen können Einfluss auf die Cytochrom-P450-Enzymaktivität haben. So ist z.B. bei CYP2C19*2 und CYP2C19*3 - Anlageträgern die CYP2C19- Aktivität eingeschränkt bis hin zum völligen Fehlen der Aktivität bei Homozygotie für diese Polymorphismen.
Ca. 15 % - 30% der Patienten weisen - je nach ethnischer Zugehörigkeit - CYP2C19-Allele mit verminderter Aktivität auf. Diese Patienten haben unter Clopidogrel-Therapie ein bis zu 3-fach höheres Risiko, ein kardiovaskuläres Ereignis zu erleiden, als Patienten mit normaler CYP2C19-Aktivität. Die FDA (Food and Drug Administration) hat bereits im März 2010 mit entsprechenden Warnhinweisen in der Packungsbeilage von Clopidogrel (Plavix) reagiert. Für betroffene Patienten kann eine Dosisanpassung oder eine andere Medikation (z.B. Prasugrel, Acetylsalicylsäure) in Erwägung gezogen werden.
Ergänzende Hinweise Arzneimittelinteraktionen:
Da zudem eine Vielzahl von Medikamenten wie z.B. die Protonenpumpeninhibitoren (PPI) ebenfalls über CYP2C19 verstoffwechselt wird, kann eine Kombination mit Clopidogrel zu erniedrigten Plasma-Konzentrationen des aktiven Metaboliten führen. Eine routinemäßige Verordnung starker oder moderater CYP2C19-Inhibitoren (Omeprazol, Esomeprazol) zusammen mit Clopidogrel ist daher zu vermeiden und Therapiealternativen sind in Erwägung zu ziehen.
Indikationen für eine CYP2C19-Genotypisierung bei Clopidogrel-Therapie
- geplanter oder erfolgter Beginn einer Clopidogrel-Therapie, insbesondere bei Patienten, die ein hohes Risiko für kardiovaskuläre Komplikationen tragen (z.B Stentpatienten). Hier sollte die Genotypisierung möglichst VOR Therapiebeginn erfolgen, da die größte Gefahr für thromboembolische Ereignisse bei Langsam-Metabolisierern in den ersten Tagen bzw. im ersten Monat besteht
- schlechtes Ansprechen der ex vivo Thrombozyten-Reaktivität auf eine Clopidogrel-Behandlung (Non-Responder, sog. „Clopidogrel-Resistenz“)
- Abklärung der Ursache von unerwünschten Ereignissen unter Clopidogrel-Therapie, z. B. Blutungen oder thrombotische kardiovaskuläre Ereignisse
Material
2 ml EDTA-Blut
Hinweis:
Ausnahmekennziffer: 32010
Schriftliche Einwilligungserklärung gemäß GenDG erforderlich
Literatur
Sibbing et al. Circulation 2010;121:512-518,
Mega et al. N Engl J Med 2009;360:354-362
Mega et al. JAMA 2011;306:2221-2228,
Scott et al. Clin Pharmacol Ther. 2011;90(2):328-332
https://www.fda.gov
Einfluss neuer direkter oraler Antikoagulanzien auf Gerinnungsanalysen
Seit 2008 wurden eine Reihe neuer Wirkstoffe eingeführt, die als neue oder direkte orale Antikoagulanzien (NOAK bzw. DOAK) bezeichnet werden, und die aufgrund ihrer kontinuierlichen Zulassungserweiterung immer breitere Anwendung finden.
Es kann zwischen zwei Wirkprinzipien unterschieden werden: zum einen der direkte Thrombin-Hemmer Dabigatran (Pradaxa®) und zum anderen die Faktor-Xa-Hemmer Rivaroxaban (Xarelto®), Apixaban (Eliquis®) und Edoxaban (Lixiana®).
Da es nur für Dabigatran mit Idarucizumab ein Antidot gibt, ergeben sich dringende Fragen zum Verfahren in Notfallsituationen und nach dem Stellenwert der in der Routine und Notfallsituation verfügbaren Gerinnungstests (1, 4).
Dabigatran hemmt direkt Thrombin (Faktor IIa) und beeinflusst deshalb viele Thrombin-basierte funktionelle Gerinnungsanalysen („Clotting-Teste“). Es führt z. B. zu einer erheblichen Verlängerung der aPTT und der Thrombinzeit. Aber auch der „Quick-Wert“ (Prothrombinzeit) kann in geringerem Maße zu niedrig, der INR zu hoch gemessen werden.
Die Faktor Xa-Inhibitoren führen zu falsch erniedrigten Quick- und erhöhten INR-Ergebnissen. In geringerem Ausmaß kann aber auch die aPTT verlängert werden. Am wenigsten beeinflusst Apixaban die Globalteste.
Je nach Messprinzip werden von diesen Medikamenten auch viele andere Gerinnungs-analysen gestört, insbesondere bei therapeutischer aber auch bei prophylaktischer Dosis (3).
Die gängigen Gerinnungsteste sind aber n i c h t ausreichend für ein Therapiemonitoring geeignet. Eine Übersicht über die zu erwartenden Effekte ist auf Seite 2 tabellarisch zusammengefasst.
Vorgehen vor operativen Eingriffen:
Aufgrund der häufigen Beeinflussung durch die DOAK ist ein noch von manchen Operateuren gewünschter „präoperativer Gerinnungsstatus“ mit Quick und aPTT bei DOAK-Patienten sinnlos, da ein „pathologisches“ Testergebnis zu erwarten ist und nur die korrekte Einnahme durch den Patienten dokumentiert.
Bei etwaigen Notfalleingriffen sprechen eine normale Thrombinzeit bei Dabigatran und normale aPTT- und Quick-Werte bei bei Dabigatran und Faktor Xa-Inhibitoren jenseits von 3-4 Stunden nach Einnahme gegen ein klinisch relevantes Blutungsrisiko (4).
Ein bei Phenprocoumon (Marcumar®) häufig notwendiges „Bridging“ (präoperative Umstellung auf Heparin) kann bei den DOAK aufgrund der kurzen Halbwertszeiten entfallen: je nach Eingriff wird das Präparat einfach 12-48 Stunden vor Eingriff abgesetzt und nach Op ebenfalls situationsabhängig nach 8-72 Stunden wieder angesetzt (4).
Hinweise zu Blutungskomplikationen sind u.a. im Deutschen Ärzteblatt aufgeführt (1, 4).
Fazit:
Viele Analysenergebnisse werden in Abhängigkeit von Untersuchungsmethode und Testreagenz in unterschiedlichem Ausmaß verfälscht. Die auftretenden Störeffekte sind daher nicht generell von Labor zu Labor vergleichbar! Grundsätzlich unbeeinflusst bleiben Analysenergebnisse, wo eine Konzentration mittels Immunoassays ermittelt wird: dies betrifft die D-Dimere und das von-Willebrand-Faktor-Antigen.
Monitoring:
Ein routinemäßiges Monitoring ist bei den DOAK nicht vorgesehen. Für diese Medikamente gibt es für Notfall- und Sondersituationen spezielle für das Monitoring geeignete Untersuch-ungen, die allerdings nicht in jedem Labor zur Verfügung stehen. Für Dabigatran steht eine modifizierte „verdünnte“ Thrombinzeit zur Verfügung. Das Monitoring der Faktor Xa-Inhibitoren basiert auf einer für das jeweilige Medikament kalibrierten Anti-Xa-Messung. Je nach Fragestellung sollte die Blutentnahme vor Einnahme des Medikaments (Talspiegel zum Ausschluss einer Kumulation/Überdosierung) oder 2-4 Stunden nach Einnahme (Peakspiegel zum Wirksamkeitsnachweis) erfolgen. Für die Untersuchungen benötigen wir Citrat-Blut, bitte notieren Sie auf dem Überweisungsschein: Dabigatran- bzw. Rivaroxaban-, Apixaban- oder Edoxaban-Monitoring.
Möglicher Einfluss von direkten oralen Antikoagulanzien (Prophylaxe) auf gängige Gerinnungsanalysen (u.a. 3):
Hinweis: das Ausmaß der Beeinflussung hängt ab vom Zeitpunkt zwischen Tabletteneinnahme und Blutentnahme (Tabellenwert entspricht Ausmaß für Peakspiegel). Zudem gibt es aber zum Teil auch ausgeprägte Unterschiede in Abhängigkeit des Testherstellers (Reagenzieneinfluß)
| Gerinnungstest | Dabigatran | Rivaroxaban | Apixaban | Edoxaban |
| aPTT | ↑/↑↑ | ↔/↑ | ↔ | ↔/↑ |
| Quick | ↓ | ↓ | ↔ | ↓ |
| INR | ↑ | ↑ | ↔ | ↑ |
| Thrombinzeit | ↑↑↑ | ↔ | ↔ | ↔ |
| Fibrinogen nach Clauss | ↔ | ↔ | ↔ | ↔ |
| Faktor VIII Clotting Test | ↓↓ | ↓ | ↓ | ↓ |
| Faktoren IX, XI, XII | ↓↓ | ↓/↓↓ | ↓ | ↓ |
| Faktoren II, V, VII, X | ↓ | ↓ | ↓ | ↓ |
| Faktor XIII (chromogen) | ↓ | ↔ | ↔ | ↔ |
| Antithrombin (chromogen über Xa) | ↔/↑ | ↑ | ↑ | ↑ |
| D-Dimer | ↔ | ↔ | ↔ | ↔ |
| von-Willebrand-Faktor-Antigen | ↔ | ↔ | ↔ | ↔ |
| von-Willebrand-Faktor-Aktivität (früher Ristocetin-Co-Faktor) | ↔ | ↔ | ↔ | ↔ |
| Protein C (chromogen) | ↔ | ↔ | ↔ | ↔ |
| Protein S Aktivität | ↑↑ | ↑↑ | ↑↑ | ↑↑ |
| APC-Resistenz | ↑ (falsch-neg.) | ↑ | ↑ | ↑ |
| Lupus-Antikoagulans | ↑↑ (falsch-pos.) | ↑↑ | ↔ | ↑↑ |
Literatur:
1. Neue direkte Orale Antikoagulanzien: Was im Notfall zu beachten ist. Steiner, T., Dtsch Ärztebl 2012; 109 (39): A-1928
2. Der Gerinnungsimpuls. Der aktuelle Ratschlag zur Gerinnungsdiagnostik; Prof. Dr. E. Lindhoff-Last , Editorial, 2012 Siemens Healthcare Diagnostics GmbH, Ausgabe 1/2012
3. Die Einflüsse von Antikoagulanzien auf Routine- und Spezialdiagnostik im Gerinnungslabor; Fr. Prof. Dr. E. Lindhoff-Last und Herrn PD Dr. D. Peetz. 2010 Roche Diagnostics
4. Direkte orale Antikoagulanzien in der traumatologischen Notaufnahme. Maegele et al. Dtsch Arztebl Int 2016; (35-36): 575-82
Fachinformationen - Humangenetik
Genetische Diagnostik bei Infertilität
Die Zahl der ungewollt kinderlosen Paare in Deutschland liegt bei ca. 15% (1). Die Fortschritte in der Reproduktionsmedizin, insbesondere seit der Einführung des direkten Mikroinjektionsverfahren von Spermien (ICSI), ermöglichen es nun auch prinzipiell einer Reihe von infertilen Paaren ihren Kinderwunsch zu erfüllen. Ist die Ursache der Infertilität allerdings eine Chromosomenaberration oder ein molekulargenetischer Defekt in einem Gen, ist eine Übertragung dieser genetischen Anomalie auf die Nachkommenschaft möglich. In der Routinediagnostik werden folgende drei zytogenetische und molekulargenetische Untersuchungstechniken von der Deutschen Gesellschaft für Reproduktionsmedizin 2004 (2) empfohlen, um Betroffene erkennen und entsprechend beraten zu können.
Zytogenetische Aberrationen bei Männern
Ca. 13,1% der Männer mit Azoospermie und ca. 4,3% der Männer mit Oligozoospermie weisen chromosomale Anomalien auf. Bei Männern mit Azoospermie können 93% der chromosomalen Anomalien auf Aberrationen der Geschlechts-chromosomen zurückgeführt werden, wobei hier 47,XXY (Klinefelter-Syndrom) dominiert. Infertilität ist bei 47,XXY Männern ohne Mosaik fast unvermeidlich. Männer mit Mosaiken XXY/XY können hingegen Spermien in variabler Anzahl produzieren. Bei Männern mit Oligozoospermie dominieren Aberrationen der autosomalen Chromosomen, wobei die Robertson - Translokationen (vor allem 13;14), gefolgt von reziproken Translokationen, überwiegen.
Die Vererbungsmuster von Robertson- Translokationen sind komplex und abhängig von den beteiligten Chromosomen und dem Geschlecht des Trägers. Im Gegensatz zu Frauen ist bei Männern mit einer Robertson-Translokation (13;14) das Risiko eines Nachkommens mit einer unbalancierten Translokation sehr gering (<1%), da die Produktion abnormer Spermatozoen wahrscheinlich selektionsbedingt gering ist.
Bei Trägern von reziproken Translokationen variiert die Wahrscheinlichkeit eines Nachkommen mit abnormalem Chromosomensatz zwischen 0 - 50% in Abhängigkeit von den beteiligten Chromosomen und der Größe der Rearrangements. Die Wahrscheinlichkeit für eine Chromosomen-anomalie ist umso größer, je geringer die Spermienzahl ist. Es besteht jedoch kein Zusammenhang zwischen Spermienmorphologie bzw. Spermienmotilität und dem Risiko für chromosomale Anomalien.
Ca. 2,5-4% der Frauen mit einer Indikation für eine IVF weisen Chromosomenaberration wie numerische gonosomale Mosaike (45,X/46,XX oder 45,X/47,XXX/46,XX etc.) oder reziproke Translokationen auf. Bei Paaren die ICSI (intracytoplasmic sperm injection) in Anspruch nehmen, weisen nicht nur die männlichen Partner (4,6%) sondern interessanterweise auch die Partnerinnen (1,8-5%) eine erhöhte Rate an Chromosomenanomalien auf.
Indikation: Infertilität unklarer Genese
Material: 10 ml unzentrifugiertes Li-Heparinblut
Methode: Karyotypanalyse nach Kurzzeit-kultivierung
Gendefekte: Mutationen im CFTR-Gen
Bei ca. 2% aller Männer mit Infertilität ohne Zeichen einer Cystischen Fibrose (CF, Mukoviszidose), bei ca. 6% der Patienten mit obstruktiver Azoospermie und bei 99% aller männlichen, adulten CF-Patienten liegt eine ein- oder beidseitige Abwesenheit der Samenstränge (Congenital bilateral absence of vas deferens, CBAVD) vor (4,5). CF ist eine der häufigsten autosomal rezessiven Erkrankungen in der kaukasischen Bevölkerung (Trägerfrequenz 1:25), welche durch Mutationen im CFTR-Gen (Cystic Fibrosis Transmembrane Regulator) hervorgerufen wird. Über 900 Mutationen des CFTR-Gens wurden beschrieben, wobei 88% der CF-Patienten
Homozygot für zwei „schwere“ Mutationen und 11% compound (=zusammengesetzt) heterozygot für eine „schwere“ und eine „milde“ Mutation sind.
Ca. 65% der Patienten mit CBAVD weisen Mutationen im CFTR-Gen auf und prägen entweder eine schwache Cystische Fibrose aus oder sind symptomfrei. Ca. 88% dieser CBAVD-Patienten sind compound heterozygot für eine „schwere“ und eine „milde“ CFTR-Gen-Mutation, 12% weisen „milde“ Mutationen auf beiden elterlichen CFTR- Allelen (Genkopien) auf. Allerdings findet man bei CBAVD andere Kombinationen von mutanten Allelen als bei CF. Neben einer compound Heterozygotie für R117H und ΔF508 besteht der häufigste, mit CBAVD assoziierte Genotyp aus dem 5T-Allel in Verbindung mit einer schwerwiegenden Mutation. Die Zahl der TG-Repeats in der Nachbarschaft eines 5T-Allels hat einen Einfluss auf die Ausprägung des Phänotyps (6).
Das Risiko einer CF für Nachkommen von (ungetesteten) CBAVD Paaren, die sich ICSI unterziehen liegt zwischen 1:100 und 1:200, wenn man davon ausgeht, dass die meisten CBAVD-Patienten eine schwere CF auslösende CFTR-Mutation besitzen und die a priori Trägerfrequenz bei den Partnerinnen je nach Herkunft bis zu 1:25 beträgt. Daher sollte bei CBAVD auch die Partnerin auf Mutationen im CFTR-Gen getestet werden.
Indikation: Männer mit CBAVD und deren Partnerin, ggf. männliche Infertilität unklarer Genese
Material: 5 ml EDTA-Blut
Methode:
Stufe 1: Sequenzanalyse der häufigsten Mutationen im CFTR-Gen (gemäß EBM)
abhängig von dem Ergebnis in Stufe 1
Stufe 2: Komplettanalyse des CFTR-Gens (MLPA und Sequenzierung)
Y-Mikrodeletionen: Mutationen in dem Azoospermiefaktor
Der auf dem Y-Chromosom gelegene Azoo-spermiefaktor (AZF) ist für die Spermatogenese von essentieller Bedeutung und wird auf der Region Yq11 in 3 Subintervalle AZFa, AZFb und AZFc unterteilt. Ca. 4-14% der Patienten mit nicht-obstruktiver Oligozoospermie und ca. 11-18% der Patienten mit Azoospermie weisen mikroskopisch nicht erfassbare Mikrodeletionen in diesen Bereichen auf (7).
Die Y-Gene im AZFa Locus sind in der Embryogenese und im Kindesalter für die Bereitstellung und Differenzierung der Spermatogonien von Bedeutung, die Y-Gene im AZFb und AZFc Locus sind für Reifung der Spermatogonien wichtig. Eine AZFa Deletion ist häufig mit einem Fehlen von Keimzellen verbunden, so dass eine testikuläre Spermienextraktion (TESE) eher erfolglos sein wird. Bei einer AZFb Mikrodeletion ist die Chance für das Auffinden von Spermien sehr gering. Bei der am häufigsten vorkommenden AZFc Mikrodeletion findet sich ein sehr variabler Phänotyp von Azoo- bis Oligozoospermie (8).
Im Falle einer AZF-Deletion sind männliche Nachkommen nach ICSI ebenfalls Träger dieser Deletion und werden mit großer Wahrscheinlichkeit ebenfalls subfertil bzw. infertil sein. Dabei ist jedoch eine mögliche variable Expressivität zu beachten.
Indikation: Infertile Männer mit nicht obstruktiver, idiopathischer Azoospermie, Oligozoospermie, Kryptospermie, Sertoli Cell Only-Syndrom, OAT (Oligoasthenoteratozoospermie)-Syndrom
Material: 5ml EDTA-Blut
Methode: DNS-Isolierung mit anschließender Amplifikation von 6 nicht-polymorphen STS- Markern aus den Regionen AZFa, AZFb und AZFc des Y-Chromosoms. Nachweis einer Deletion mittels Gelelektrophorese
Zytogenetische Aberrationen bei Frauen
Chromosomenstörungen bei der Frau finden sich bei ca. 3,3-9,8 % aller infertilen Paare. Als häufigste Aberration ist das Ullrich-Turner-Syndrom (45,X) zu nennen. Bis zu 50% der Trägerinnen zeigen Mosaikbefunde unter anderem mit strukturellen Aberrationen des X- Chromosoms (39%), mit einem derivativen Y-Chromosom (6%) oder mit numerischen Aberrationen (7%). Bei einigen Mosaikbefunden (z.B. mos 45,X/46,XX) kann es zur Keimzellbildung kommen. Da das Risiko für eine prämature Ovarialinsuffizienz (POI) bei Betroffenen erhöht ist, sollte eine rechtzeitige Realisierung eines Kinderwunsches empfohlen werden, beziehungsweise die Abklärung der ovariellen Reserve und gegebenenfalls Asservierung von Eizellen. Im Fall eines Mosaiks mit Anteilen des Y-Chromosoms unter Beteiligung des SRY-Gens ist ein erhöhtes Gonadoblastom-Risiko zu berücksichtigen.
Bei Frauen mit 47,XXX-Karyotyps (Inzidenz etwa 1:1000) ist die Fertilität in jungen Jahren gegeben, doch liegt ein erhöhtes Risiko für eine POI vor.
Bei infertilen/subfertilen Frauen werden auch vermehrt strukturelle Chromosomenaberrationen gefunden, allerdings seltener als bei infertilen Männern. Betreffen sie das X-Chromosom, können sie ein erhöhtes Risiko für eine POI bedingen.
Das Risiko für chromosomal kranke Kinder bei elterlichen strukturellen Chromosomenve-ränderungen muss je nach Art der Chromosomen-aberration beurteilt werden. Bei gonosomalen numerischen Aberrationen liegt in der Regel kein gegenüber der Durchschnittsbevölkerung erhöhtes Wiederholungsrisiko vor.
Indikation: Infertilität unklarer Genese
Material: 5 ml unzentrifugiertes Heparinblut
Methode: Karyotypisierung nach Kurzzeitkultivierung
Gendiagnostik: FMR1 (Fragile X mental Retardation 1)-Gen
Als Ursache für einen frühen Eintritt der Menopause vor dem 40. Lebensjahr findet sich bei ca. 10-15% der betroffenen Frauen eine Verlängerung eines Triplettrepeats im FMR1-Gen im Sinn einer Prämutation. In der Eizellreifung kann es zur weiteren Verlängerung des CGG-Triplettrepeats in den Bereich der Vollmutation kommen. Dieses Gen ist ursächlich für das Fragile-X-Syndrom (fra(X)-Syndrom), der häufigsten monogen bedingten Ursache einer Entwicklungs-verzögerung und mentalen Retardierung. Aufgrund der X-chromosomalen Vererbung findet sich das Vollbild der Erkrankung bei betroffenen Jungen und Männern, seltener bei Mädchen und Frauen.
FMR1-Prämutationsträgerinnen haben ein Risiko von ca. 20% für eine POI. Außerdem ist das Risiko für ein „Fragile X Related Tremor/Ataxia Syndrome“(FXTAS) erhöht. Prämutations-trägerinnen haben ein erhöhtes Risiko für Nachkommen mit einer mentalen Retardierung (fra(X)-Syndrom).
Indikation: Frauen mit Fertilitätsstörungen und erhöhten FSH-Werten vor dem 40. Lebensjahr oder POI/Menopause bei zwei Familienmitgliedern bei unauffälligem Karyogramm
Material: 10 ml EDTA-Blut
Methode: DNA-Amplifikation (PCR) des variablen Bereichs im Exon 1 des FMR1-Gens zum Nachweis einer CGG-Expansion, Fragmentlängenanalyse mittels Kapillarelektrophorese, gegebenenfalls Southern Blot nach EcoRI / EagI-Restriktionsverdau
21-Hydroxylase-Defizienz (CYP21A2-Gen)
Beim Adrenogenitalen Syndrom (AGS) handelt es sich um eine autosomal-rezessiv vererbte Erkrankung, die überwiegend durch Mutationen im 21-Hydroxylase-Gen (CYP21A2) auf Chromosom 6p21.3 verursacht wird. Klinisch unterscheidet man das klassische kongenitale AGS von der late-onset Form (Prävalenz 1:1000), wobei sich Letzteres bei erwachsenen Frauen vor allem durch polyzystische Ovarien, Zyklusstörungen und Oligo-Amenorrhoe, Hirsutismus und Akne manifestieren kann. Verursacht wird das late-onset AGS durch Homozygotie einer "milden" Mutation oder durch kombinierte (compound) Heterozygotie einer "milden" und einer "schweren" Mutation im 21-Hydroxylase-Gen.
Indikation: Verdacht auf 21-Hydroxylase-Defizienz (late-onset Form)
Material: 5 ml EDTA Blut
Methode: Sequenzierung des CYP21A2-Gens, Multiplex Ligation-dependent Probe Amplifikation (MLPA)
Empfehlung: Im Fall einer Homozygotie oder Compound-Heterozygotie sollte unbedingt der Partner untersucht werden, da die Heterozygotenfrequenz in Zentraleuropa bei 1:50 liegt. Erhält ein Kind von beiden Elternteilen eine „schwere“ Mutation, kann es an der klassischen Form eines kongenitalen AGS erkranken. Bei spezifischen Fertilitätsstörungen oder Syndromverdacht können weitere gezielte Analysen angezeigt sein.
Weiterführende laboratoriumsmedizinische Differentialdiagnostik
- Spermiogramm
- Endokrine Störungen: Testosteron, Östradiol, DHT, LH, FSH, TSH, Prolaktin
- Ggf. bakteriologische und virologische Abklärung
- Antikörper gegen Spermien
Begriffserklärungen
Unauffälliger Chromosomensatz: 44 Autosomen und 2 Gonosomen (Karyotyp 46,XY bzw. 46,XX)
Numerische Chromosomenanomalien: die Zahl betreffend
Strukturelle Chromosomenanomalien: Umbauten innerhalb eines Chromosoms oder zwischen verschiedenen Chromosomen.
- Balanciert: ohne Verlust und/oder Zugewinn von chromosomalem Material;
->meist keine klinische Symptomatik - Unbalanciert: mit Verlust und/oder Zugewinn;
->Ursache einer syndromalen Erkrankung - Reziproke Translokation: wechselseitiger Austausch von Chromosomensegmenten ohne erkennbaren Materialverlust.
- Robertson Translokation: Fusion der langen Arme zweier akrozentrischen Chromosomen mit Verlust der kurzen Arme. Da die kurzen Arme keine Einzelgene enthalten, bedingt der Verlust keine phänotypischen Veränderungen; z. B.„erbliche“ Trisomie 21, Sterilität.
Literatur:
1.) Bruckert E.; How frequent is unintentional childlessness in Germany?; Andrologia, 1991, 23 (3): 245-50
2.) Ludwig M. et al.: Stellungnahme der Arbeitsgemeinschaft Reproduktionsgenetik der Deutschen Gesellschaft für Reproduktionsmedizin: Empfehlungen zur genetischen Diagnostik bei Kinderwunschpaaren. J Reproduktionsmed. Endokrinol, 2004, 1(3): 190-3
3.) Mau U.A.: Somatic chromosomal abnormalities in infertile men and women. Cytogenet Genome Res, 2005, 111: 317-336
4.) Jarzabek K. et al.: Cystic fibrosis as a cause of infertility. Reproductive Biology, 2004, 4 (2): 119 – 129
5.) Dohle G. R. et al.: The complex relationships between cystic fibrosis and congenital bilateral absence of the vas deferens: clinical, electrophysiological and genetic data. Human Reproduction, 1999, 14: 371-374
6.) Cuppens H. et al.: CFTR mutations and polymorphisms in male infertility. Int J Androl, 2004, 27: 251-256
7.) Vogt P. H. : Azoospermia factor (AZF) in Yq11: towards a molecular understanding of its function for human male fertility and spermatogenesis. Reproducitve BioMedizine Online, 2005, 10 (1): 81-93
8.) Foresta C. et al.: Y Chromosome micordeletions and alterations of spermatogenesis. Endocr Rev , 2001, 22: 226-239
Array-CGH bei unklarem Dysmorphie- und/oder Retardierungssyndrom
Die Array-CGH, auch molekulare Karyotypisierung genannt, ist eine Methode zur Erweiterung der herkömmlichen zytogenetischen Diagnostik. Chromosomale Veränderungen (lmbalancen), die mit der konventionellen zytogenetischen Auswertung nicht erfasst werden können, lassen sich mit dieser Methode nachweisen.
In der zytogenetischen Analyse im Lichtmikroskop sind chromosomale Stückverluste (Deletionen) und Zugewinne (Duplikationen) in der Regel erst ab ca. 5- 10 Megabasen (Mb, 1Mb = 1000 kb) zu erkennen. Die Array-CGH erlaubt einen Nachweis von lmbalancen mit einer extrem hohen Auflösung (unter 50 Kilobasen (kb)).
Solche lmbalancen treten nicht selten auf. Einige sind relativ häufig und bereits gut beschrieben, wie zum Beispiel das DiGeorge-Syndrom oder das Williams Beuren-Syndrom. Mittlerweile wurden zahlreiche weitere chromosomale lmbalancen nachgewiesen, die als neue Mikrodeletions- und -duplikations- Syndrome definiert werden konnten.
Bei Kindern mit mentaler Retardierung und unauffälliger konventioneller Chromosomenanalyse findet sich zu etwa 10-20% eine solche lmbalance, die den Phänotyp hinreichend erklärt.
Indikation:
Voraussetzung für eine Array-CGH ist eine vorab durchgeführte konventionelle Chromosomenanalyse, deren Ergebnis die diagnostische Fragestellung nicht hinreichend beantworten konnte. Eine ergänzende Array-CGH Untersuchung ist bei Patienten mit Verdacht auf eine syndromale Erkrankung sinnvoll, bei denen eine oder mehrere der folgenden Symptome ohne Hinweis auf eine bekannte monogene Erkrankung vorliegen:
- mentale Retardierung,
- Entwicklungsstörungen aus dem Autismus-Formenkreis,
- Entwicklungsverzögerung
- Fehlbildungen und Funktionsstörungen des Gehirns unklarer Ursache,
- Wachstumsstörungen,
- angeborene Fehlbildungen / Dysmorphien.
Bei Patienten mit einer unspezifischen syndromalen Erkrankung kann die Array-CGH auch zur näheren Abklärung und Bruchpunktbestimmung von zytogenetisch erkennbaren Chromosomenanomalien, z. B. Deletionen, Duplikationen, Markerchromosomen oder balanciert erscheinender Strukturveränderungen verwendet werden.
Methodik:
Für die Diagnostik wird ein hochauflösender Oligo Array mit ca. 180.000 Oligo-Sonden verwendet („DNA Chip“).
Etwa gleiche Mengen genomischer Patienten DNA und Referenz-DNA werden mit unterschiedlichen Fluorochromen markiert und gemeinsam auf dem Array hybridisiert (CGH = „comparative genomic hybridization“). Durch Messung der lnten-sitätsverhältnisse der verwendeten Fluoreszenz-farbstoffe sind Dosisunterschiede, also Deletionen und Duplikationen, nachweisbar. Bei dem verwendeten Chip liegt die Nachweisgrenze für lmbalancen bei etwa 20 kb.
Beurteilung:
Der Nachweis einer lmbalance mit möglicher Relevanz für die Symptomatik des Patienten wird, soweit dies möglich ist, durch Fluoreszenz in situ Hybridisierung (FISH) mit einer entsprechenden Sonde bestätigt. In manchen Fällen wird auch eine Abklärung über eine Elternuntersuchung durchgeführt. Die Beurteilung der Pathogenität wird zum einen anhand der Position und Frage nach klinisch relevanten Genen, die in dem deletierten oder duplizierten Bereich liegen, beurteilt. Zum anderen wird in den international verfügbaren Datenbanken geprüft, ob bereits Patienten mit der gefundenen Aberration beschrieben wurden und wenn ja, welche Symptomatik vorliegt. Die diagnostizierte Aberration kann de novo vorliegen, nicht selten werden Mikrodeletionen oder - duplikationen jedoch von einem Elternteil vererbt. Der Phänotyp kann oftmals auch innerhalb einer Familie sehr unterschiedlich sein.
Material:
Array-CGH: 2 ml EDTA-Vollblut (separates Röhrchen)
Karyotypisierung/FISH: 5 ml Heparin-Blut
Laut Gendiagnostikgesetz muss eine schriftliche Einwilligungserklärung der Patientin / des Patienten, bzw. des Erziehungsberechtigten vorliegen.
Zytogenetische Diagnostik bei habitueller Abortneigung
Ca. 10-15 % aller klinischen Schwangerschaften enden als spontane klinische Aborte, meist als Frühaborte vor Beendigung der 12. Schwangerschaftswoche (SSW) (1). Bei Einbeziehen von den meist unerkannten sehr frühen Aborten muss man von einer Abortrate von ca. 50% ausgehen (1). Die Wahrscheinlichkeit eines Abortes steigt dabei mit der Zahl vorausgegangener Fehlgeburten (1). Von allen Frauen mit Kinderwunsch leiden etwa 1-3% unter habituellen Aborten (1).
Unter den zahlreichen Ursachen (anatomisch, autoimmunologisch, endokrinologisch, hämostaseologisch und exogen) sind die chromosomal bedingten am häufigsten.
Die zytogenetische Abklärung von Aborten wird aus klinischen, therapeutischen, prognostischen und nicht zuletzt auch aus psychologischen Gründen von der Deutschen Gesellschaft für Gynäkologie und Geburtshilfe 2004 (2) empfohlen.
Elterliche Chromosomenaberrationen
Chromosomenanalysen aus Blut-Lymphozyten von Paaren mit habituellen Aborten zeigen in ca. 5% der Fälle Auffälligkeiten (3). Dabei überwiegen balancierte reziproke Translokationen bei Mann oder Frau, durch die unbalancierte Zustände bei Fruchtanlagen entstehen können (3). Der Nachweis dieser Chromosomenaberration bei den betroffenen Paaren ist 30fach höher als in der Allgemeinbevölkerung (3). Andere Auffälligkeiten sind die elterliche Robertson Translokation, sowie strukturelle oder numerische Aberrationen der Geschlechts-chromosomen (z.B. Klinefelter-Mosaik) (3). Je nach Art der diagnostizierten Strukturaberration bei den Betroffenen kann dann das Risiko eines erneuten Abortes abgeschätzt werden.
Chromosomenaberrationen in Spontanaborten
Untersuchungen an frühen Spontanaborten haben ergeben, dass in 50-60% eine Chromosomenaberration vorliegt, wobei numerische Chromosomenanomalien überwiegen (Tabelle 1).
| Chromosomaler Befund | Spontanaborte |
| Autosomale Trisomien v. a. Trisomie 13, 16, 18, 21, 22 | Ca. 50 - 60 % |
| Polyploidie | Ca. 20 % |
| Monosomie X (Ullrich-Turner Syndrom) | Ca. 15 % |
| Strukturelle Aberrationen | Ca. 5% |
Tabelle 1: % Verteilung von Chromosomenanomalien bei Spontanaborten mit Chromosomenaberrationen. (Modifiziert nach 4,5,6)
Wiederholungsrisiko
Maternales Altersrisiko: Mit steigendem Alter der Mutter ist das Risiko von Chromosomenfehlverteilungen z.B. Trisomie 21 bei Nachkommen erhöht (7). Daher ist mit zunehmendem mütterlichen Alter auch mit einer Zunahme der Frühaborte zu rechnen.
Numerische Chromosomenaberrationen in Aborten entstehen überwiegend spontan während der Reifeteilung der Eizelle oder des Spermiums. Für die überwiegende Mehrheit der betroffenen Paare (> 95%) ist hier das Wiederholungsrisiko nur geringfügig erhöht. Ein kleiner Teil der Paare (< 5%) mit Trisomie-Befunden bei Aborten weist jedoch Keimzellmosaike auf, welche in somatischen Zellen nicht nachzuweisen sind. Ein Keimzellmosaik ist anzunehmen, wenn bei normalem Chromosomensatz beider Partner wiederholt die gleiche Chromosomenanomalie im Abortgewebe nachgewiesen wird (8). Das Wiederholungsrisiko ist für die Betroffenen je nach Anteil der trisomen Keimzellen in den Gonaden eventuell erheblich erhöht.
Die relativ seltenen strukturellen Chromosomenaberrationen bei Aborten entstanden zur Hälfte der Fälle de novo, das heißt neu, und lagen zur anderen Hälfte bereits bei einem Elternteil als balancierte „erbliche“ bzw. familiäre Strukturaberration vor. Bei einer neu entstandenen Strukturaberration ist das Wiederholungsrisiko sehr gering und besteht im Wesentlichen in der Möglichkeit eines Gonadenmosaiks.
Das Risiko für einen unbalancierten Chromosomensatz des Feten bei familiär bekannten Chromosomen-aberrationen (z.B. Translokation, Inversion) beträgt in Abhängigkeit von der Art der Aberration sowie der Lokalisation und Größe des beteiligten Chromosomenabschnittes ca. 0 - 22% (9, 10). Bei Iso-Translokation, wie z.B. (13;13) oder (21;21) kommt es in nahezu 100% der Fruchtanlagen zu einem unbalancierten Chromosomensatz.
Spätaborte weisen aufgrund der natürlichen Selektion von Feten mit chromosomalen Aberrationen bezogen auf strukturelle Aberrationen meist einen normalen Chromosomensatz auf (1). Eine numerische Aberration, wie zum Beispiel Trisomie 18, kann als Ursache eines Spätaborts vorliegen.
Aufgrund der relativ großen Häufigkeit spontan auftretender fetaler Chromosomenanomalien bei den Abortursachen ist ein unauffälliger Karyotyp in einer vorangegangenen Fehlgeburt eher mit einem erhöhten Risiko für einen erneuten Abort assoziiert (11). Hier sollte gezielt nach anderen Ursachen wie z. B. endokrinologischen Auffälligkeiten, Autoimmunerkrankungen und hereditären Thrombophilien (s. u.) gesucht werden.
Begriffserklärungen
Unauffälliger Chromosomensatz: 44 Autosomen und 2 Gonosomen (Karyotyp 46,XY bzw. 46,XX)
Numerische Chromosomenanomalien: die Zahl betreffend
- Trisomie: zusätzliches Chromosom vorhanden, z.B 47,XY,+21
- Polyploidie: ganzzahlige Vermehrung haploider Chromosomensätze auf z. B. 69 bzw. 92 Chromosomen
Strukturelle Chromosomenanomalien: Umbauten innerhalb eines Chromosoms oder zwischen verschiedenen Chromosomen.
- Balanciert: ohne Verlust und/oder Zugewinn von chromosomalem Material;
->meist keine klinische Symptomatik - Unbalanciert: mit Verlust und/oder Zugewinn;
->Ursache einer syndromalen Erkrankung - Reziproke Translokation: wechselseitiger Austausch von Chromosomensegmenten ohne erkennbaren Materialverlust.
- Robertson Translokation: Fusion der langen Arme zweier akrozentrischer Chromosomen mit Verlust der kurzen Arme. Da die kurzen Arme keine Gene enthalten, bedingt der Verlust keine phänotypischen Veränderungen; Ursache für „erbliche“ Trisomie 13 oder 21, Sterilität
- Inversion: Eine Inversion entsteht durch zwei Brüche in einem Chromosom mit einer 180°-Drehung des Segmentes zwischen den Bruchstellen.
Chromosomales Mosaik: Vorhandensein verschiedener Chromosomensätze innerhalb von oder in unterschiedlichen Organsystemen
Zytogenetische Untersuchungen
Chromosomenanalyse aus Abortmaterial
- Indikation: vorangegangene Aborte, bekannte elterliche Chromosomenveränderungen, vorangegangene Geburten von Kindern mit Chromosomenveränderungen
- Material: Chorionzotten oder fetales Gewebe in Transportmedium
- Methode: Karyotypanalyse nach Langzeitkultur
Chromosomenanalyse aus peripheren Lymphozyten
- Indikation: Sterilität, Geburten von Kindern mit chromosomalen Anomalien, habituelle Aborte, auffälliger pränataler Chromosomensatz beim ungeborenen Kind, V. a. auf Dysmorphie-Syndrom, V. a. gonosomale Chromosomenveränderungen, Verwandte von Personen mit strukturellen Chromosomenanomalien
- Material: 5 ml unzentrifugiertes Heparinblut
- Methode: Karyotypanalyse nach Kurzzeitkultivierung
Weiterführende laboratoriumsmedizinische Differentialdiagnostik
- Thrombophilien: Antiphospholipid-Syndrom (Lupusantikoagulans, Anti-Cardiolipin-Ak), Protein C-, Protein-S-Mangel, Faktor-V-Leiden, MTHFR-, Prothrombinmutation, AT III, Homocystein, Faktor VIII, IX, XII
- Endokrine Störungen: FSH, LH, TSH, Progesteron, Östradiol, Prolactin, Testosteron, DHEAS, Androstendion
- Ausschluss eines Diabetes mellitus: oraler Glukosetoleranztest, Nüchternglukose, HbA1c
- Mikrobiologischer Abstrich der Cervix
- Umweltbedingte Ursachen: Quecksilber, Blei, Cadmium, Pestizide, organ. Lösungsmittel, Kupfer, Schwefeldioxid
- Immunologische Ursachen: Bestimmung von NK-Zellen im peripheren Blut, Bestimmung der TH2/TH1-Ratio
Literatur
- DGGG, Leitlinien, Empfehlungen, Stellungnahmen, Stellungnahme zur Diagnostik und Therapie des wiederholten Spontanabortes, 2004, 3.2.8
- Fryns J. et al.; Structural chromosome rearrangements in couples with recurrent fetal wastage; Eur J Obstet Gynecol Reprod Biol, 1998, 171-176
- Ohno M. et al.; A cytogenetic study of spontaneous abortions with direct analysis of chorionic vili ; Obstet Gynecol, 1991, 77(3) : 394-8
- Gardo S. et al.; Cytogenetic analysis of spontaneous abortions with direct analysis of chorionic villi; Eur J Obstet Gynecol Reprod Biol, 1992, 47: 117-20
- Eiben B. et al.; Cytogenetic analysis of 750 spontaneous abortions with the direct-preparation method of chorionic villi and its implications for studying genetic causes of pregnancy wastage; Am J Hum Genet, 1990, 47 (4): 656-63
- Snijders RJ. Et al.; Maternal age- and gestation-specific risk for trisomy 21; Ultrasound Obstet Gynecol, 1999, 13 (3): 167-70
- Tseng LH. et al.; Recurrent Down`s syndrome due to maternal ovarian trisomy 21 mosaicism; Arch Gynecol Obstet, 1994; 255 (4): 213- 6
- Midro AT. et al.; Experiences with risk estimates for carriers of chromosomal reciprocal translocations; Clin Genet, 1992, 41(3): 113-22
- Barisic I. et al.; Risk extimates for balanced reciprocal translocation carriers-prenatal diagnosis experience ; Clin Genet, 1996, 49 (3): 145-51
- Ogasawara M. et al.; Embryonic karyotype of abortuses in relation to the number of previous miscarriages; Fertil Steril, 2000, 73 (2): 300 – 304
Hämochromatose
Die hereditäre Hämochromatose (HH) Typ 1 ist mit einer Prävalenz von 2-5/1000 die häufigste angeborene, möglicherweise lebensbedrohliche Stoffwechsel-Erkrankung in Populationen keltischer Abstammung (Skandinavien, Nord-Europa, Groß-Britannien). Bei den Betroffenen kommt es durch eine erhöhte Eisenresorption im Dünndarm sukzessive zur Eisenanreicherung in verschiedenen Organen, insbesondere in der Leber. Klinische Symptome treten zumeist zwischen dem 4. und 6. Lebensjahrzehnt auf, wobei Männer i.d.R. stärker betroffen sind als Frauen. Bei frühzeitiger Diagnose ist die Erkrankung gut therapierbar, unbehandelt führt sie häufig zu Leberzirrhose, Lebertumoren, Diabetes mellitus und/oder Herzrhythmusstörungen.
Genetik:
Die Vererbung der hereditären Hämochromatose Typ 1 erfolgt autosomal-rezessiv. Im entsprechenden HFE-Gen findet sich bei 85-90% der Betroffenen eine Homozygotie für den C282Y-Polymorphismus, bis zu 6% der europäischen Fälle sind mit einer C282Y/H63D Compound-Heterozygotie assoziiert. Die überwiegende Mehrheit der C282Y Homozygoten entwickelt im Laufe des Lebens auffällige Eisenserumparametre bis hin zu einer klinisch manifesten Hämochromatose, wobei die Angaben zur genauen Penetranz in der Literatur stark schwanken. Für C282Y/H63D Compound-Heterozygote wird die Erkrankungswahrscheinlichkeit mit 1-2 % angegeben, für H63D Homozygote ist diese gering. Bei Homozygotie für einen weiteren, seltenen S65C-Polymorphismus oder kombinierte Heterozygotien für C282Y/S65C und H63D/S65C wird nur ein leicht erhöhtes Erkrankungsrisiko gefunden.
Insgesamt sind mehr als 20 verschiedene HFE Kandidatenmutationen mit heriditärer Hämochromatose Typ 1 assoziiert, deren genaue klinische Bedeutung und Prävalenz ist jedoch nicht in allen Fällen bekannt ist.
Vorgehen:
Um irreversible Spätschäden zu vermeiden ist eine möglichst frühe Diagnosestellung anzustreben. Hauptsymptome der Hämochromatose sind eine Lebervergrößerung, das Auftreten eines Diabetes mellitus sowie eine dunkle Hautpigmentierung. Oft wird bei Betroffenen aber auch als Erstes eine Herzrhythmusstörung auffällig. Allgemeinere Symptome wie starke Müdigkeit, Reizbarkeit, depressive Verstimmung, Infektanfälligkeit, nachlassende Libido und atypische Gelenkbeschwerden oder eine Hypothyreose können ebenfalls auf eine Hämochromatose hindeuten.
Zur weiteren Abklärung empfiehlt sich die Laborbestimmung von Serumferritin, Transferrinsättigung und Serumeisen. Bei auffälligen Befunden (z.B. Transferrinsättigung > 45 %, erhöhtes Ferritin, typischerweise > 1000 µg/l, Serum Fe > 30 µmol/l) ist eine molekulargenetische Untersuchung des HFE-Gens (insbesondere C282Y, H63D) indiziert.
Bei klinisch gesicherter Hämochromatose ist darüber hinaus eine weitergehende Mutationsanalyse gerechtfertigt, wenn aufgrund des ethnischen Hintergrunds der Verdacht auf Vorliegen einer anderen Mutation besteht, oder wenn bei einem Patienten mit Heterozygotie für C282Y weitere Faktoren (wie z.B. Transferrinsättigung > 50% und Ferritinwert > 200 µg/l; positive Leberbiopsie) auf eine hereditäre Hämochromatose hindeuten bzw. generell, wenn das Vorliegen einer hereditären Hämochromatose durch eine Leberbiopsie bestätigt wurde.
Ist eine Mutation im HFE-Gen nachgewiesen worden, sollten auch alle Verwandten zumindestens ersten Grades auf den Defekt hin untersucht werden.
Therapeutisch lässt sich eine Hämochromatose durch wiederholte Aderlässe behandeln, wobei der Ferritinspiegel in der Erhaltungsphase in einem Bereich von 20-50 µg/l liegen sollte. Bei asymptomatischen Anlageträgern sind regelmäßige Kontrolluntersuchungen der Transferrin-Sättigung und des Ferritins indiziert.
Hinweis:
Die molekulargenetische Diagnostik erfolgt aus 2-5 ml EDTA-Blut.
Nach Gendiagnostikgesetz benötigen wir eine Einverständniserklärung des Patienten.
Ausnahmekennziffer: 32010
Literatur
Bacon et al. Diagnosis and Management of Hemochromatosis: 2011 Practice Guidelines by the American Association for the Study of Liver Diseases. Hepatology. 2011; 54(1): 328–343. Aguilar-Martinez et al. Iron overload in HFE C282Y heterozygotes at first genetic testing: a strategy for identifying rare HFE variants. Haematologica. 2011;96(4):507-14 Gabriel and Stuhrmann-Spangenberg, medgen 18 (2006) Leitlinien zur molekulargenetischen Diagnostik der Hereditären Hämochromatose, Stuhrmann et al., Clinical utility gene card for: Haemochromatosis [HFE] Eur J Hum Genet. 2010; 18(9) Rochette et al. Multicentric origin of hemochromatosis gene (HFE) mutations. Am J Hum Genet 1999, 64(4): 1052-1062
Laktoseintoleranz
10% bis 20% der erwachsenen Bevölkerung in Deutschland leiden an einer Laktoseintoleranz. Bei Betroffenen führt der Genuss von Milch oder Milchprodukten zu abdominellen Beschwerden wie Durchfall, Blähungen und Bauchschmerzen. Ursächlich ist eine verminderte Aktivität des Bürstensaumenzyms Laktase, welches Laktose nur unzureichend in Glukose und Galaktose spaltet. Eine Malabsorption von Laktose ist die Folge. Osmotische Effekte sowie die bakterielle Verstoffwechslung der Laktose im Dickdarm können zu Diarrhöen und vermehrten Bildung von Darmgasen mit entsprechenden Oberbauchbeschwerden führen.
Die Laktoseintoleranz kann auf einer ererbten oder erworbenen Störung des Laktose-Stoffwechsels beruhen.
- Die mit Abstand häufigste Ursache des Laktasemangels ist die adult-onset Form welche auf einem Polymorphismus in Position -13910 (g.-13910C>T) in der regulatorischen Region des LCT-Gens beruht.
Ca. 25% der mittel- und nordeuropäischen Bevölkerung sind homozygote Träger des inaktivierenden C-Allels und entwickeln typischerweise zwischen dem 5. und 20. Lebensjahr eine Laktoseintoleranz. Je nach ethnischer Zugehörigkeit kann dieser Prozentsatz auch wesentlich höher liegen und beträgt z.B. bei Asiaten bis zu 100 %.
- Differentialdiagnostisch hiervon abzugrenzen sind die sekundär bedingte Formen des Laktase-Mangel, welcher eine Schädigung des Darmepithels durch eine intestinale Erkrankung wie z.B. die einheimische Sprue oder auch Gastroenteritiden mit konsekutiv meist vorübergehender Einschränkung der Laktaseaktivität zugrunde liegt.
- Die seltene angeborene kongenitale Laktoseintoleranz hingegen ist eine Erkrankung des Säuglingsalters / Kleinkindesalters, welchedurch unbeherrschbare Durchfälleund Gedeihstörungen charakterisiert ist. Ursächlich hierfür sind Mutationen innerhalb des LCT-Gen, so dass ihre molekulargenetische Untersuchung entsprechend beauftragt werden muss.
Anamnestisch lässt sich das Vorliegen einer Laktoseintoleranz meist nicht hinreichend erfassen, da sich einerseits ein Teil der Patienten des Zusammenhanges zwischen der Aufnahme von Milchprodukten und dem Auftreten von Beschwerden nicht bewusst ist, andererseits aber auch ein Teil der Patienten ohne Laktoseintoleranz von Ihrer selbst gestellten Diagnose überzeugt sind.
Zur differentialdiagnostischen Abklärung trägt der Nachweis der Mutation im Promotorbereich des Gens bei, insbesondere, wenn der Laktose-Toleranztest nicht durchführbar ist.
Da eine Laktoseintoleranz eine verminderte Kalziumaufnahme nach sich ziehen kann, ist prinzipiell bei Betroffenen auf eine ausreichende Calcium und Vitamin D-Aufnahme zu achten. Dies gilt insbesondere für Kindern und Jugendlichen im Wachstum und für ältere Menschen (hier vor allem Frauen) wegen des Osteoporoserisikos.
Hinweis:
Die molekulargenetische Diagnostik erfolgt aus 2-5 ml EDTA-Blut.
Nach Gendiagnostikgesetz benötigen wir eine Einverständniserklärung des Patienten.
Ausnahmekennziffer: 32010
Literatur
Brannon PM et al.: NIH Consensus Develop ment Conference Statement: Lactose Intolerance and Health. NIH Consens State Sei Statements 2010; 27 (2): 1-27.
Bernardes-Silva CF et al.: Lactase persistence/non-persistence variants, C/T_13910 and G/A_22018, as a diagnostic tool for lactose intolerance in IBS patients. Clin Chim Acta 2007; 386: 7-11.
Keller J, Laktoseintoleranz: Der aktuelle Kenntnisstand zu Diagnostik und Therapie Arzneiverordnung in der Praxis - Band 38 • Ausgabe 4 • Juli 2011, Hamburg
Layer P, Andresen V, Pehl C et al.: S3-Leitlinie Reizdarmsyndrom: Definition, Pathophysiologie, Diagnostik und Therapie. Gemeinsame Leitlinie der Deutschen Gesellschaft für Verdauungs- und Stoffwechselkrankheiten (DGVS) und der Deutschen Gesellschaft für Neurogastroenterologie und Motilität (DGNM). Z Gastroenterol 2011; 49: 237-293
Obermayer-Pietsch,B.M. et al. Genetic predisposition for adult lactose intolerance and relation to diet, bone density, and bone fractures. J. Bone Miner. Res.19, 42 (2004)
Stellenwert vom HLA-DQ2 und HLA-DQ8 in der Zöliakie-Diagnostik
Pathophysiologie:
Die Zöliakie (Synonyme: Sprue oder Glutensensitive Enteropathie) ist eine immunologisch vermittelte Enteropathie bei genetischer Disposition mit Ausbildung immunologischer Marker. Zur Bestimmung der Zöliakie- spezifischen Antikörper stehen die Endomysium-Ak und Gewebstransglutaminase-Ak sowie Ak gegen deamidierte Gliadinpeptide (ehemals Gliadin-Ak) zur Verfügung.
Verursacht wird sie durch das Klebereiweiß Gluten in Getreidearten (Weizen, Roggen, Gerste; Hafer noch unklar). Es handelt sich hierbei um eine immunvermittelte, systemische Erkrankung, bei der es nach Aufnahme von Gluten zur Schädigung der Dünndarmschleimhaut kommt. Unter glutenfreier Kost regeneriert die Dünndarmschleimhaut und klinische Symptome sowie Zöliakie-spezifische Antikörper bilden sich zurück.
Die Zöliakie ist eine Krankheit, die sich in jedem Alter manifestieren kann. Dabei können die Betroffenen intestinale (Blähbauch, Durchfall, Krämpfe, Flatulenz, Gewichtsverlust, Schwäche und Müdigkeit, etc.) wie auch extraintestinale (Anämie, reduzierte Knochendichte, Unfruchtbarkeit, Entzündung der Mundschleimhaut, dermatologische und neurologische Störungen, etc.) Symptome zeigen. Neben symptomatischen sind auch asymptomatische Verläufe möglich.
Die Prävalenz der Zöliakie liegt bei cirka 0,2-1%. Ungefähr 5 – 20 % der Verwandten ersten Grades sind betroffen.
Einzige kausale Therapie ist die lebenslange glutenfreie Ernährung.
Genetik:
Die Zöliakie hat eine starke genetische Komponente. Personen mit genetischer Prädisposition tragen bestimmte HLA Klasse II Allele, die für die Moleküle DQ2 und DQ8 kodieren. Bei mehr als 97 % der Zöliakiepatienten sind diese Allele nachweisbar. Dabei tragen 95% der Betroffenen das DQ2-Heterodimer, die restlichen tragen meist das sogenannte DQ8- Heterodimer.
Sind diese Allele nicht vorhanden, ist es sehr unwahrscheinlich, dass eine Person an Zöliakie leidet. Träger dieser Allele müssen jedoch nicht zwangsläufig an einer Zöliakie erkranken, da 30 - 40 % der gesunden Bevölkerung ebenfalls Genträger sind. Eine HLA-Typisierung auf DQ2 und DQ8 eignet sich somit gut zur Ausschlussdiagnostik. Bei positivem Nachweis ist ggf. die Bestimmung Zöliakie-spezifischer Antikörper indiziert.
Indikation:
- Diskrepante serologische und histologische Befunde, wie bspw. negative Zöliakie-spezifische-Antikörper bei gleichzeitig geringen infiltrativen Veränderungen in den proximalen Dünndarmbiopsien
- eventuell zur Vermeidung von Biopsien bei Kindern: starker klinischer Verdacht auf Zöliakie bei gleichzeitig hochpositiven Gewebstrans-glutaminase-Ak und positivem Nachweis End-omysium Ak.
- asymptomatische Personen mit Zöliakie-assoziierten Erkrankungen: Familienmitglieder 1. Grades, Typ1 Diabetes mellitus, selektiver IgA-Mangel, Autoimmun-thyreoiditis, etc.
Diagnostik:
Molekulargenetischer Nachweis von HLA - DQ2, - DQ8 bei V.a. Zoeliakie
Material:
separates 2 ml EDTA-Vollblut-Röhrchen bzw. DNA-Probe
Laut Gendiagnostikgesetz muss eine schriftliche Einwilligungserklärung des Patienten/ der Patientin vorliegen.
Literatur
Husby S et al., ESPGHAN Guidelines for the Diagnosis of Coeliac Disease, JPGN 2012, 54(1): 136 – 160
Hourigan. Clin Exp Med 2006; 6:53–59
Farrell et al. N Engl J Med 2002; 346(3):180-188
HLA-Genotypisierung (DQB1*06:02 - Nachweis) bei Narkolepsie
Allgemeines
Unter Narkolepsie versteht man eine Schlafstörung (Dyssomnie), die durch einen Verlust von Neuronen im Hypothalamus verursacht wird. Meist in der Adoleszenz oder im jungen Erwachsenenalter beginnend, kommt es zu einer Störung im Schlaf-Wach-Rhythmus. Typisch ist dabei eine erhöhte Schläfrigkeit am Tag, wobei es zu plötzlichem "Schlafzwang" kommen kann. Der Patient fällt dabei, anders als beim Nachtschlaf, sehr schnell in einen REM-Schlaf. Der Nachtschlaf selbst ist gestört und es kann zu einer sog. "Schlaflähmung" (Kataplexie) kommen.
Pathogenese
In genetischen Studien konnte gezeigt werden, dass bei über 98% aller Narkolepsie-Patienten im HLA-System das Allel DQB1*06:02 vorhanden ist. Bei Europäern liegt dieses Allel meist im sog. DR15-DQ6-Haplotyp vor, der die Allele DRB1*15:01 - DQA1*01:02 - DQB1*06:02 umfasst. Dieser Haplotyp kommt bei Nord- und Mitteleuropäern mit einer Frequenz von 14 bis 20 % vor. Es wird jedoch davon ausgegangen, dass die Prävalenz nur etwa 0,05 % beträgt. Dies bedeutet, dass die Erkrankung nur relativ selten zum Ausbruch kommt, es gibt aber vermutlich eine hohe Anzahl von Patienten mit Narkolepsie, bei denen die Symptomatik als Schlaflosigkeit (Insomnie), Depression, Epilepsie oder Drogenabusus diagnostiziert wird.
Klinische Bedeutung
Der molekulargenetische Nachweis erfolgt durch eine Polymerase-Kettenreaktion (PCR), bei der mit Hilfe von sequenz- bzw. allelspezifischen Primern definierte Fragmente der Allele DRB1*15:01 - DQA1*01:02 - DQB1*06:02 amplifiziert bzw. nicht amplifiziert werden. Fehlen diese Allele (insbesondere DQB1*06:02), kann eine Narkolepsie mit hoher Wahrscheinlichkeit ausgeschlossen werden. Dagegen bedeutet der Nachweis des Allels DQB1*06:02 nicht, dass der Patient auch tatsächlich an Narkolepsie erkrankt ist oder erkranken wird.
Anforderung:
HLA-Genotypisierung (DQB1*06:02) bei V.a. Narkolepsie
Einverständniserklärung des Patienten nach GenDG nötig
Material:
2 - 3 ml EDTA-Blut
Literatur:
Mignot et al. - Am J Hum Genet 68, 686-699 (2001)
Fachinformationen - Infektiologie - Mikrobiologie
Hepatitis D und E: eine unterschätzte Gefahr !?
Hintergrund:
Bei der Differenzialdiagose der viralen infektiösen Hepatitis standen bisher die Hepatitis A, B und C im Vordergrund. Die Hepatitis D (Delta) und E galten in Deutschland als Seltenheit, die allenfalls bei bestimmten Risikogruppen in Betracht gezogen wurde. Neue künftige Behandlungs-möglichkeiten bei der Hepatitis D und die Zunahme von in Deutschland erworbenen Hepatitis E-Infektionen haben in jüngster Zeit die Aufmerksamkeit auf diese verkannten „Stiefkinder“ der Hepatologie gelenkt.
Hepatitis D
Das Hepatitis D- oder Hepatitis-Delta-Virus (HDV) ist ein inkomplettes RNA-Virus, das zu seiner Replikation die Hülle des Hepatitis B-Virus benötigt (HBs-Ag) und daher nur zusammen mit HBV (Koinfektion) oder auf einen chron. HBV-Träger übertragbar ist (Superinfektion). Das Virus wird parenteral übertragen, vertikale und sexuelle Übertragungen sind seltener als bei HBV. Bei der Superinfektion kommt es häufig zu schweren, nicht selten fulminanten Verläufen, aber auch die Koinfektion verläuft schwerer als die Monoinfektion mit HBV. In 70-90 % der Fälle kommt es zu schweren chronischen Verläufen. Die Erkrankung ist in Deutschland sicher unterdiagnostiziert, die Zahl der Infizierten wird aber auf etwa 50.000 geschätzt. Man rechnet damit, daß bei uns etwa 10% aller Hepatitis B-Erkrankten auch mit dem HDV infiziert sind. Die Therapie mit pegyliertem Interferon α führt nur bei etwa einem Viertel der Patienten zu einer andauernden HDV-Eliminierung, Rückfälle können noch nach Jahren auftreten. Künftige Therapieansätze befinden sich noch in einem frühen Forschungsstadium.
Hepatitis E
Das Hepatitis E-Virus (HEV) wird im Stuhl ausgeschieden und fäkal-oral übertragen. Wichtigste Ansteckungsquelle sind konta-miniertes Trinkwasser oder Lebensmittel, selten enger Kontakt mit Infizierten. Es gibt 8 Genotypen, wobei in Deutschland der Genotyp 3 vorherrscht. Die Mehrzahl der Infektionen verläuft asymptomatisch, seltener als akute Virushepatitis mit einer Letalität von bis zu 1%, bei Schwangeren allerdings bis zu 20%, falls sie sich in Asien oder Afrika mit dem Genotyp 1 infizieren. Chronische Verläufe werden bei den in Europa vorkommenden Genotypen 3 und 4 beschrieben bei immunsupprimierten Patienten (Transplantatempfänger, HIV/AIDS, Chemotherapie). HEV galt bisher als exotische Reiseinfektion bei Reisen nach Asien, Afrika sowie Mexico. Neueren Daten zufolge werden allerdings aktuell 90 % der an das RKI gemeldeten Fälle in Deutschland erworben! Laut RKI ist sie bei uns mittlerweile die häufigste virale Hepatitis mit geschätzt 400.000 Infektion pro Jahr. Bei deutschen Blutspendern findet man bei etwa 17 % HEV-Antikörper, was auf eine Vielzahl asymptomatischer Verläufe hindeutet. Als wichtigste Infektionsquelle hierzulande wurde unzureichend gegartes Fleisch oder Rohwürste von Schwein, Wildschwein und Hirsch identifiziert, bei denen das HEV weit verbreitet ist. Weitere mögliche Übertragungswege sind Bluttransfusionen und Organtransplantationen. Ein Impfstoff ist in China zugelassen, steht in Europa aber nicht zur Verfügung.
Indikationen zur Testung:
- HDV-Ak (IgG und IgM): bei jeder akuten oder chronischen Hepatitis B (HBs-Ag und/oder HBV-DNA positiv). Wenn positiv auch Bestimmung der HDV-RNA mittels PCR.
- HEV-Ak (IgG und IgM): bei Hepatitis im Rahmen der Stufendiagnostik nach Ausschluß von Hepatitis A, B und C auch ohne Reiseanamnese. Der Nachweis von HEV-RNA im Blut oder Stuhl beweist eine akute Infektion.
Material
Serum, EDTA-Blut (für HDV- und HEV-RNA)
Abrechnung:
Die Kosten der serologischen Antikörper-Tests werden von den gesetzlichen und privaten Krankenversicherungen übernommen.
Meldepflicht:
Für alle Hepatitiden A, B, C, D und E besteht Meldepflicht nach § 6 und § 7 des IfSG.
Literatur
- RKI-Ratgeber Hepatitis B und D (2016)
- Manns MP. Hepatitis D (Delta) – die vergessene Herausforderung. Arzneimittelforschung 2010
- Faber et al. Hepatitis E Virus Seroprevalence among Adults, Germany. Emerg Inf Dis 2012
- RKI-Ratgeber Hepatitis E (2015)
- Faber et al. Case-control study on risk factors for acute hepatitis E in Germany, 2012 to 2014. Euro Surveill 2018
Norovirus-Nachweis aus Stuhl
Erreger
Der Norovirus (NV), gehört zur Familie der Caliciviren. Sie sind weltweit verbreitet und für einen Großteil der nicht bakteriell bedingten infektiösen Gastroenteritiden verantwortlich. Der Mensch ist das einzige bekannte Reservoir des Erregers.
Die Erkrankung tritt im gesamten Jahresverlauf auf, jedoch mit einer deutlichen saisonalen Häufung ab Mitte Oktober bis ins zeitige Frühjahr.
Übertragung
Die Übertragung erfolgt vorwiegend über direkten Kontakt von Mensch zu Mensch als Schmierinfektion (Handkontakt mit kontaminierten Flächen), fäkal-oral (Stuhl, Erbrochenes), aerogen durch die orale Aufnahme virushaltiger Tröpfchen, die im Rahmen des schwallartigen Erbrechens entstehen, über Lebens-mittel und auch über das Trinkwasser. Die erforder-liche Infektionsdosis liegt bei weniger als 100 Virus-partikeln. Die Meldedaten der letzten Jahre bestätigen, dass Kinder unter 5 Jahren und ältere Personen über 70 Jahre besonders häufig betroffen sind. Folglich ist der Norovirus die überwiegende Ursache von Ausbrüchen akuter Gastroenteritis in Gemeinschaftseinrichtungen, Krankenhäusern, Altenheimen etc., aber auch Ursache sporadischer Erkrankungen. Bei Säuglingen und Kleinkindern stellen sie nach den Rotaviren die zweithäufigste Ursache akuter Gastroenteritis dar.
Hygienemaßnahmen
Wichtig ist die Einhaltung der allg. Hygienemaß-nahmen in Gemeinschaftseinrichtungen und Küchen. Insbesondere während der symptomatischen Phase müssen die Maßnahmen ausgeweitet werden:
konsequente Händehygiene und Händedesinfektion, Absonderung der erkrankten Personen, Tragen von Handschuhen, Schutzkittel, ggf. Mundschutz, Desinfektion von patientennahen Flächen, Toiletten, Waschbecken, Türgriffen.
Zur Desinfektion sind nur Präparate mit nachgewiesener viruzider Wirksamkeit geeignet (Angaben des Herstellers beachten).
In Privathaushalten muss ebenfalls auf eine sorgfältige Hände- und Toilettenhygiene geachtet werden (Hände und Flächen regelmäßig und gründlich reinigen), Kontakt zu Erkrankten möglichst meiden, Reinigung kontaminierter Flächen und Gegenstände (empfohlen werden die Benutzung von Haushaltsgummi-handschuhen und Einwegtüchern, die dann entsorgt werden können, um die Viren nicht weiter zu verbreiten.
Klinik
Die IKZ beträgt 6-50 Stunden.
Eine durch NV verursachte Gastroenteritis äußert sich zumeist durch starke Übelkeit, heftiges, schwallartiges Erbrechen und schweren Durchfall. Die Symptome können bis zu 3 Tagen anhalten. Die Viren werden i.d.R. 7-14 Tage (selten bis vier Wochen) über den Stuhl ausgeschieden.
Wegen der ausgeprägten Variabilität der NV und dem Fehlen einer umfassenden protektiven Immunität sind Reinfektionen möglich, eine Impfung steht nicht zur Verfügung.
Die Therapie erfolgt symptomatisch durch Ausgleichen des Flüssigkeits- und Elektrolytverlustes, ggf. Antiemetika.
Diagnostik
Der Nachweis bei uns im Labor erfolgt durch einen ELISA–Test aus dem Stuhl, der die Typen 1 + 2 erfaßt.
Es steht auch eine PCR zur Verfügung die die Diagnostik ergänzen kann. Die Kosten übernimmt im Falle eines negativen ELISA-Testes die GKV.
Abrechnung
Da der Keim lt. IfSG vom 01. 01. 2001 meldepflichtig ist, können Sie die budgetbefreiende Ausnahmeziffer 32006 auf den Überweisungsschein der Kassenpatienten schreiben.
Für Rückfragen stehen wir Ihnen gern jederzeit zur Verfügung.
Lit.: 1. RKI Ratgeber Infektionskrankheiten – Merkblätter für
Ärzte;
2. HygMed 2007;32 (1/2)
Mycobacterium tuberculosis (MTB) IFN-release assay (IGRA): QuantiFERON-TB-Gold-Test aus Blut
In Deutschland und West-Europa ist die Tuberkulose(TB)-Inzidenz derzeit niedrig, es kommen jedoch immer wieder Einzelfälle vor. In Osteuropa, Afrika und Südostasien sind hingegen die Tuberkulose-Fallzahlen nach wie vor hoch und weiter zunehmend. Die TB ist ferner bei immunsuppressiver Therapie und im Zusammenhang mit Infektionen durch Humane Immundefizienz-Viren (HIV) von besonderer Relevanz.
Zum Screening und zur Diagnosestellung der TB steht seit langer Zeit der Tuberkulin-Hauttest (Mendel-Mantoux bzw. Tine-Test) zur Verfügung. Falsch positive Ergebnisse dieses Tests (z.B. durch BCG-Impfung, Infektion mit atypischen Mykobakterien (MOTT)) und falsch negative Ergebnisse (immunsupprimierte Patienten, HIV-Infektion) schränken die Anwendung dieses Tests jedoch stark ein.
Neue Tests: TB Interferon-γ-release assays
Es stehen zwei neue, einander ähnliche Verfahren (IGRAs) zur Verfügung. Gegenüber den Tuberkulin-Hauttests zeichnen sie sich durch eine erhöhte Spezifität (> 95%) für den M. tuberculosis-Komplex (M. tuberculosis, M. bovis. M. africanus) aus, sie reagieren praktisch nicht auf BCG und MOTT. (Zum Vergleich: Spezifität des Tuberkulin-Tests 40 - 70%). Auch die Sensitivität der IGRAs liegt bei > 90% und wird durch den Immunstatus des Patienten kaum beeinflusst.
Für einen IGRA werden die T-Lymphozyten des Patienten mit Antigenen von M. tuberculosis inkubiert. Hatte das Immunsystem Kontakt mit Tuberkelbakterien, reagieren die T-Zellen mit der Produktion von Interferon-γ, welches anschließend durch eine Immunfärbung nachgewiesen wird
Indikationen:
- mit chronisch entzündlichen, immunmodulierenden Erkrankungen vor der Erstgabe von TNF-a-Blockern
- mit einer HI-Virus-Infektion nur vor einer Therapieentscheidung einer behandlungsbedürftigen Infektion mit M. tuberculosis-Komplex (außer BCG)
- vor Durchführung einer Organtransplantation (Niere, Herz, Lunge, Leber, Pankreas)
Ferner Einsatz durch den ÖGD zum Screening auf TB (Risikogruppen, z.B. nach Kontakt mit TB-Patienten), durch Arbeitsmedizin, sowie in Kliniken ergänzend zur klinischen Diagnosestellung TB.
Vorteile gegenüber Tuberkulin-Hauttests:
- geringe Zahl falsch positiver Ergebnisse (kaum Reaktion auf BCG-Impfung oder MOTT)
- höhere Sensitivität bei immunsupprimierten Pat.(u.a. HIV-Infektion!), bei denen der Tuberkulin-Hauttest negativ ausfällt, und bei Kindern
- geringerer Aufwand am Patienten
- Ausschluss von Fehlern beim Hauttest-Ablesen
Einschränkung:
- keine Unterscheidung zwischen latenter, aktiver und abgelaufener/therapierter TB
Anforderung:
auf den Ü-Schein bitte schreiben: „TB-QuantiFERON-Test“
Material:
ein Lithium-Heparin-Röhrchen (vorher bei uns bestellen) korrekt gefüllt, nach Blutentnahme 8-10 mal schwenken! Röhrchen am Entnahmetag ins Labor! Vor Frost und Hitzeeinwirkung schützen, in Styropor-Box verpackt. Nicht im Kühlschrank lagern!
Präanalytik:
Ist hier besonders wichtig, da lebende Zellen gebraucht werden.
Nicht im Kühlschrank lagern! Nicht der direkten Sonnenstrahlung aussetzen!
Separate Verpackung beim Transport ins Labor, am besten in extra Styropor-Box (vom Labor mit den Röhrchen zusammen anfordern) bei Zimmertemperatur dem Laborfahrer extra geben und/oder großen Zettel „nicht kühlen“ anbringen.
Literatur:
1. Deutsches Zentralkomittee zur Bekämpfung der Tuberkulose
(www.pneumologie.de)
2. Pai M, et al. Lancet Infect Dis 2004: 4; 761-76
3. Chapman et al. AIDS 2002: 16; 2285-93
4. Diel et al. Chest 2009: 135; 1010-8
5.,6. Leitlinien Fachges. Dermatologie, Rheumatologie
Fachinformationen - Klinische Chemie
Diagnostik des primären Hyperaldosteronismus mittels Aldosteron-Renin Quotient
Häufigste sekundäre Ursache der arteriellen Hypertonie ist der primäre Hyperaldosteronismus (PHA), Conn-Syndrom. Betroffen sind neueren Studien zufolge 5 - 12 % aller Hypertoniker. Dabei hat sich herausgestellt, dass bis zu 90 % der Patienten normokalämisch sind. Das Fehlen einer Hypokaliämie schließt einen primären Hyperaldosteronismus also nicht aus!
Allgemeines
Ein PHA wird in mehr als 90 % durch ein Aldosteron-produzierendes Adenom oder durch eine idiopathische beidseitige Nebennieren-hyperplasie ausgelöst.
Aldosteron ist ein Mineralkortikoid und wird in der Nebennierenrinde gebildet. Aldosteron bewirkt eine Wasser- und Natriumretention. Die Aldosteronseretion unterliegt dem Renin-Angiotensin-Aldosteron-System, einem Schlüssel-mechanismus der Blutdruckregulation.
Als bester Screening-Test gilt derzeit die Bestimmung des Aldosteron-Renin-Quotienten (ARR). Beim PHA ist die normale Abhängigkeit des Aldosterons vom Renin aufgehoben und Aldosteron disproportional in Relation zum Renin erhöht. Weiterhin wird die Reninsekretion durch die von Aldosteron induzierte renale Natrium- Resorption gehemmt.
Ein ARQ > 20 ist als potentiell pathologisch zu betrachten (Sensitivität 89%, Spezifität 96%).
Die Aldosteron-Konzentration muss dabei als zusätzliches Kriterium berücksichtigt werden, beim Conn-Syndrom liegt sie im oberen Normbereich oder darüber. Bei einem pathologischern ARQ ist ein Bestätigungstest angezeigt (z.B. Kochsalzbelastungstest).
Vorteile
Der Aldosteron-Renin-Quotient ist ein Parameter, der im Gegensatz zur Bestimmung der Einzelparameter nicht durch salzhaltige Nahrung oder Abnahmebedingungen (Körperposition) beeinflusst wird.
Indikationen
- Therapieresistente Hypertonie (mindestens 3 Medikamente und RR > 140/90)
- junge Hypertoniker (< 40 J)
- Hypokaliämische Hypertonie
- Zufällig entdeckter Nebennierentumor und Hypertonie
Präanalytik
Blutabnahme morgens nach 10 minütiger Ruhepause am sitzenden Patienten, Nüchternheit nicht notwendig.
Ein Pausieren antihypertensiver Medikamente ist notwendig bei:
| 1 Woche | ß-Rezeptoren-Blocker midazolinrezeptorantagonisten (z.B. Clonidin) Schleifendiuretika Angiotensin-II-Antagonisten |
| 4 Wochen | Spironolacton Eplerenon Drospirenon |
Erlaubt sind:
Thiaziddiuretika, ACE-Hemmer, α-Blocker, Calciumantagonisten
Material
1 ml gefror. EDTA-Plasma und 1 ml Serum.
Bei Anforderung von Aldosteron, Renin und ARQ wird der Aldosteron-Renin-Quotient aus unseren Ergebnissen berechnet.
Literatur:
Diederich et al., Med Klin 2007; 102:16-22
Schirpenbach et al., Dtsch Arztebl 2009; 106(18):305-11
Antikörper gegen Cyclisches citrulliniertes Peptid (CCP-AK)
Allgemeines
Die rheumatoide Arthritis (RA) betrifft ca. 1% der erwachsenen Bevölkerung und hat eine Inzidenzrate von ca. 30 Fällen pro 100.000 Einwohnern. In der Frühphase der Erkrankung ist die Diagnose oft nicht einfach zu stellen. Die Bestimmung des Rheumafaktors ist nicht immer hilfreich, da es sowohl RA-Fälle ohne Rheumafaktor-Nachweis also auch andere Erkrankungen wie Kollagenosen und Infektionen gibt, die mit einem positiven Rheumafaktor-Nachweis einhergehen können. Einen Fortschritt in der Labordiagnostik stellen nun die Antikörper gegen das Cyclische citrullinierte Peptid dar. Diese Antikörper richten sich insbesondere gegen Citrullin, eine seltene Aminosäure, die durch das Enzym Peptidylarginindesaminase aus Arginin entsteht. Citrullin ist wesentlicher Bestandteil des Proteins Filaggrin, das Keratinfilamente miteinander verknüpft. Citrulliniertes Fibrin wurde auch in der Synovia von Rheumapatienten gefunden. Durch die Citrullinierung verliert sich bei prädisponierten Patienten die Immuntoleranz gegen die betroffenen Proteine.
Patienten mit rheumatoider Arthritis weisen meist schon im Frühstadium der Erkrankung AK gegen Cyclisches citrulliniertes Peptid auf. Die Spezifität der CCP-AK (Zweitgeneration) liegt bei 95 – 99%, die Sensivität bei 60 – 88% und ist vergleichbar mit der Rheumafaktor-Bestimmung. Die Kombination von CCP-AK und Rheumafaktor erhöht die Sicherheit zur Diagnose der rheumatoiden Arthritis.
Sind beide Marker positiv, erhöht sich der positive prädiktive Wert auf 98%. Sind beide Marker negativ, beträgt der negative prädiktive Wert 92,5%.
CCP-AK haben somit eine hohe positive Aussagekraft für die Entwicklung einer rheumatoiden Arthritis. Somit stellen diese Antikörper eine wertvolle Hilfe dar, um frühzeitig mit einer antirheumatischen Therapie beginnen zu können.
Indikation
V.a. rheumatoide Arthritis, DD der Kollagenosen
Material
1ml Serum
Abrechnungshinweis
Bitte prüfen Sie, ob Sie die Ausnahmekennziffer 32023 ansetzen können.
Literatur:
Genth: Rheumatoide Arthritis, JlabMed 26(2002) 130
Vincent et al.: Detection of Antibodies to Deiminated Recombinant rat Filsggrin ba Enzymlinked-Immunosorbent-Assay, Arthritis Rheum 46 (2002) 2051
-Labor und Diagnose; 7. Auflage
Cystatin C
Chronische Nierenerkrankungen mit Einschränkung der glomerulären Filtrationsrate und drohender Dialyse sind häufige Erkrankungen in Praxis und Klinik.
Zur Beurteilung der glomerulären Filtrationsrate werden gewöhnlich das Serum-Kreatinin und die Kreatinin-Clearance herangezogen.
Die Serum-Kreatininwerte sind abhängig von der Muskelmasse (zu berücksichtigen bei Kindern, Frauen, schmächtigen Menschen, Akromegalie, erhöhter Muskelmasse) und können durch die Nahrung beeinflusst werden (hoher Fleischkonsum). Die Serum-Kreatininwerte steigen erst bei einer Verminderung der GFR um 50 % über die Norm an (kreatininblinder Bereich). Die Sammeluringewinnung kann ungenau und unzuverlässig sein (Kinder, Pflegepatienten). Insgesamt zeigt sich für das Serum-Kreatinin eine inadäquate diagnostische Sensitivität.
Cystatin C ist ein idealer Marker zur frühzeitigen und sicheren Beurteilung einer renalen, auch subklinischen, Dysfunktion. Cystatin C ist nicht nahrungs- und geschlechtsabhängig. Es wird endogen in konstanter Rate gebildet, glomerulär frei filtriert, tubulär weder reabsorbiert, noch sezerniert und auch nicht extrarenal eliminiert. Cystatin C ist ein empfindlicherer Marker als Kreatinin zur Erkennung leichter Einschränkungen der GFR. Das betrifft insbesondere den Kreatinin-blinden Bereich der GFR von 80-50 [ml/min/1,73 m2].
Im Vergleich zu Serum-Kreatinin ist Cystatin C der sensitivere und spezifischere Parameter für die Beurteilung der GFR und deshalb als Screening-Parameter besser geeignet als das Kreatinin.
Dies gilt besonders bei:
- Kindern und hochbetagten Patienten
- Patienten mit Typ II Diabetes
- Patienten mit akuter oder chronischer renaler Dysfunktion, z.B. bei SLE/ Autoimmuner-krankungen/HUS
- Hämodialyse/Nierentransplantation
- Steroid-Therapie oder anderer potentieller nierentoxischer Medikation
Indikation:
Beurteilung der Nierenfunktion
Material:
0,5 ml Serum
Literarur:
- Thomas und Diagnose; 7. Auflage
Procalcitonin - effektiver Einsatz von Antibiotika in der Praxis
Allgemeines
PCT ist eine Vorstufe des Hormons Calcitonin. Während Calcitonin nur in der Schilddrüse gebildet wird, wird PCT bei entsprechendem Stimulus einer bakteriellen Infektion vor allem in parenchymatösen Organen gebildet und in die Blutbahn abgegeben.
Der PCT-Spiegel steigt infolge einer bakteriellen Infektion deutlich an. Bei Virus- und Autoimmunerkrankungen oder allergischer Reaktion steigt der PCT-Spiegel nur wenig oder gar nicht an.
Kinetik:
Bereits 2–3 Stunden nach Induktion des ent-zündlichen Geschehens steigt der PCT-Spiegel an und erreicht maximale Werte nach 24 Stunden. Danach nimmt der Spiegel wieder ab (prognostisch günstig) oder verharrt auf hohen Werten bzw. steigt weiter an (ungünstige Prognose). Um den klinischen Verlauf einer Infektion/Sepsis zu beurteilen, sollte PCT wiederholt bestimmt werden. Falsch erhöhte Werte werden u.a. nach schwerer Operation, schwerem Trauma, schweren Verbrennungen, calcitoninproduzierenden Tumoren gemessen.
Mit dem Wert erhalten Sie eine individuelle Interpretation des Befundes, diese muss selbstverständlich immer im Zusammenhang mit Anamnese und Krankheitsverlauf gesehen werden.
Mehrere Studien belegen inzwischen, dass mit Hilfe der Untersuchung des PCT-Spiegels die Entscheidung zur Gabe von Antibiotika verbessert werden kann. Bei gleichen Therapieerfolgen kann in ca. 50% der Fälle das Antibiotikum eingespart werden. Außerdem kann die Häufigkeit des Antibiotika-Einsatzes und die Menge der verordneten Antibiotika wesentlich verringert werden.
Indikation
PCT hat Bedeutung als Labormarker zur frühzeitigen Erkennung einer durch Bakterien ausgelösten Infektion bzw. Sepsis. Bei tiefen Atemwegsinfektionen ist PCT geeignet, eine begründete Entscheidung für oder gegen eine Antibiotika-Therapie zu treffen. Außerdem ist der PCT-Spiegel geeignet, den Infektionsverlauf zu überwachen. Grundsätzlich gilt:
<0,25ng/ml bakterielle Infektion unwahrscheinlich
>0,25ng/ml bakterielle Inf. wahrscheinlich, Anti-biotikagabe empfohlen/indiziert.
Rund 75% der ambulant verordneten Antibiotika werden bei Atemwegsinfektionen verschrieben.
Etwa 80% aller akuten Atemwegsinfektionen sind aber nicht bakteriell verursacht. Trotzdem wird in der Befürchtung, dass sich die Viruserkrankung durch eine zusätzliche bakterielle Infektion verkomplizieren könnte, ein Antibiotikum verschrieben. Andererseits werden Antibiotika manchmal auch zu spät eingesetzt, was zu lebensbedrohlichen Situationen führen kann.
Die Messung des PCT kann bei tiefen Atemwegsinfektionen (akute Bronchitis, Exazerbation einer chronischen Bronchitis, Pneumonie) die initiale Antibiotikaverschreibung und die Dauer der Antibiotikagabe deutlich reduzieren. Das gilt sowohl für den stationären als auch für den ambulanten Bereich der Hausarztpraxis.
Ein traditioneller Labormarker mit ähnlicher Indikation ist das C-reaktive Protein (CRP). Im Vergleich des diagnostischen Werts der beiden Biomarker erlaubt das PCT ein deutlich rascheres Erkennen des entzündlichen Prozesses sowie seiner Schwere und darüber hinaus eine bessere Unterscheidung zwischen bakterieller und viraler Infektion.
Material
Serumröhrchen
Abrechnungshinweis
Sie können für die Laboranforderung Procalcitonin die im April 2018 neu von der KBV eingeführte Ausnahmekennziffer 32004 ansetzen und somit Ihr Laborbudget entlasten.
Ethylglucuronid – Marker für Alkoholkonsum
Der Nachweis eines zurückliegenden Alkohol-konsums kann aus verschiedenen Gründen notwendig werden: Kontrolle der Compliance bei Alkoholentzug, betriebsärztliche Untersuchungen oder forensische Fragestellungen. Dabei ist zwischen dem kurzfristigen, direkten Nachweis von Alkoholkonsum und der längerfristigen, indirekten Bestätigung eines chronischen Alkoholabusus zu unterscheiden.
Zum Beweis einer Aufnahme von Alkohol nur wenige Stunden zuvor ist die Bestimmung des Ethanolspiegels im Blut geeignet. Jedoch wird Ethanol schnell abgebaut und es können dementsprechend nur sehr kurzfristig Aussagen getroffen werden.
Chronischer Alkoholabusus hat indirekte Auswirkungen, die mittels MCV (Mean cellular volume), Erhöhung der Transaminasen (ins-besondere Gamma-GT) und CDT (Carbohydrate Deficient Transferrin) erfasst werden. Jedoch gibt es eine Vielzahl von Störfaktoren, die das Ergebnis beeinflussen können (Lebererkrankungen, Cholestase, Anämien, genetische Transferrin-Varianten usw.). Auch muss ein chronischer Alkoholabusus über mindestens einen Monat vorliegen.
Die Bestimmung von Ethylglucuronid im Urin schließt das diagnostische Fenster zwischen dem direkten Nachweis von Ethanol und den indirekten Markern des chronischen Abusus. Ethylglucuronid wird aus einem geringen Teil des aufgenommenen Ethanols gebildet und über die Niere ausgeschieden. Die Ausscheidung erfolgt deutlich langsamer als der Abbau von Ethanol, was zu einem verlängerten Zeitfenster für den Nachweis einer Alkoholaufnahme führt.
Je nach Ausmaß des Konsums ist Ethyl-glucuronid im Urin für bis zu 80 Stunden nachweisbar, mindestens jedoch für 24 Stunden. Im Serum ist der Zeitraum wesentlich kürzer. Auch geringer Alkoholkonsum (1 Glas Bier oder Wein) schlägt sich bereits in erhöhten Ethylglucuronid-Werten nieder.
Es besteht keine genaue Korrelation zwischen der Höhe der Ethylglucuronid-Ausscheidung im Urin und der konsumierten Alkoholmenge, jedoch ist eine ungefähre Abschätzung der aufgenommenen Ethanolmenge möglich, wobei aber auch die Zeitdauer bzw. der Zeitpunkt des Konsums berücksichtigt werden muss.
Eine Ethylglucuronid-Konzentration von > 5 mg/l im Serum spricht für einen Alkoholkonsum mit Ethanol-Spitzenkonzentrationen von mindestens 1,6 ‰ im Blut.
Mittels Ethylglucuronid-Bestimmung kann nicht unterschieden werden, ob es sich um einen einmaligen oder um einen chronischen Konsum handelt. Daher ist bei V.a. chronischen Alkoholabusus auch die Bestimmung von CDT indiziert.
Material:
2 ml Spontanurin
1m Serum
Methode: LC-MS-MS
Referenzbereich:
< 100 µg/l (Urin)
< 500 µg/l (Serum)
Literatur:
1. Borucki K, et al.: Detection of Ethanol Intake With New Markers. Alcohol Clin Exp Res 2005:29;781-787.
2. Alt A, et al.: Bestimmung von Ethylglucoronid in Urinproben mit internem Standard d5-Ethylglucuronid. Blutalkohol 1997:24;360-365.
Labordiagnostik bei Patienten mit Depression
Medizinischer Hintergrund
Die Depression gehört zu den häufigsten Erkrankungen überhaupt. Nach Unter-suchungen aus dem Jahr 2010 liegt in Europa die 12-Monats-Prävalenz bei 6,9% (1). Eine wichtige Aufgabe im Rahmen der fachärztlichen Abklärung einer Depression ist die Erkennung und Abgrenzung einer somatischen Erkrankung.
Klinische Bedeutung
Patienten mit Depression sprechen zu einem Teil (ca. 15-25%) auf die initiale Behandlung nicht an, da Diagnostik oder Therapie nicht angemessen durchgeführt werden. Die Ursachen dieser sog. Pseudotherapieresistenz sind vielfältig und umfassen u.a. unerkannte somatische Erkrankungen. Um mit der Vielzahl der in Frage kommenden Differentialdiagnosen besser umgehen zu können, bietet sich ein abgestuftes labordiagnostisches Vorgehen an. Sinnvolle Diagnosestufen wurden 2014 in einem Fortbildungsbeitrag des Deutschen Ärzteblattes vorgestellt (1). Wichtige Laborparameter umfassen demnach Blutbild, Elektrolyte, Trans-aminasen, Kreatinin, CRP und TSH in der ersten Stufe sowie Lues-Serologie (TPHA), Vitamin B12, Folsäure, Urinstatus, HIV-Antikörpertest, ggf. Liquor-Untersuchungen und Antidepressiva-Spiegel zur Über-prüfung der Compliance in der zweiten Stufe (1). Darüber hinaus sollte bei jedem neu diagnostizierten Patienten ein Drogenkonsum ausgeschlossen werden sowie bei Frauen im gebärfähigen Alter das Ergebnis eines Schwangerschaftstests vorliegen (2).
Material
| Großes Blutbild | EDTA-Blut |
| Elektrolyte (Na, K, Ca) | Serum |
| Transaminasen (ALAT, ASAT) | Serum |
| Kreatinin | Serum |
| CRP | Serum |
| TSH | Serum |
| Lues-Suchtest (TPHA) | Serum |
| Vitamin B12 | Serum |
| Folsäure | Serum |
| Urinstatus | Urin |
| HIV 1/2-Ak/Ag | Serum |
| SS-Test (ß-HCG) | Serum |
| Medikamentenspiegel | i.d.R. Serum |
| Drogenscreening (Amphetamine, Benzodiazepine, Canna-binoide, Cocain, Methadon/EDDP, Opiate, Buprenorphin) | Urin |
Abrechnung
Die Kosten der genannten Unter-suchungen werden von allen gesetzlichen und privaten Krankenkassen übernommen.
Literatur
1. Tom Bschor, Michael Bauer, Mazda Adli. Chronisch und therapieresistente Depression – Diagnostik und Stufentherapie. Deutsches Ärzteblatt 2014, 45:766-776.
2. Smith FA, Levenson JL, Stern TA. Psychiatric assessment and consultation. In: The American Psychiatric Publishing Textbook of Psychosomatic Medicine: Psychiatric Care of the Medically Ill, Second Edition, Levenson JL. (Ed), American Psychiatric Publishing, Inc, Washington, DC 2011. p.3.
Fachinformationen - Onkologie - Tumormarker
Molekularbiologischer Nachweis der BCR-ABL-Genrearrangements bei Leukämien
Allgemeines:
Die CML ist eine klonale Erkrankung der pluripotenten hämatopoetischen Stammzelle, die in 90-95% der Patienten durch eine charakteristische Translokation t(9; 22) gekennzeichnet ist. Diese findet sich auch bei ca. 20 - 30% der Erwachsenen bzw. 5% der Kinder mit einer ALL und so gut wie nie bei Patienten mit einer AML.
Pathogenese:
Die reziproke Translokation (=Austausch eines Bruchstückes) erfolgt zwischen Chromosom 9 und 22. Das Chromosom 22 wird dann Philadelphia Chromosom (Ph) genannt. Durch die Umlagerung des auf Chr. 9 lokalisierten ABL-Gens (ABL-Tyrosinkinase) in die auf Chr. 22 gelegene Region des BCR-Gens (BCR= Breakpoint Cluster Region) resultiert ein BCR-ABL-Fusionsgen. Dies führt zur Bildung einer hyperaktiven Tyrosinkinase, die eine ungehemmte Proliferation in den hämatopoetischen Vorläuferzellen induziert. 5-10% der Patienten mit CML weisen variante Philadelphia - Translokationen oder einen unauffälligen Chromosomensatz auf. Bei Patienten mit CML und zytogenetisch normalem Karyotyp können, vermutlich aufgrund submikroskopischer Insertionen, BCR-ABL-Transkripte nachgewiesen werden.
Abb. 1.: Entstehung des Ph-Chromosoms durch reziproke Translokation. In Abhängigkeit von der Bruchstelle innerhalb des Wildtyp-BCR-Gens, Major-bcr (M-bcr) oder minor-bcr (m-bcr) entstehen die beiden CML-typischen Fusionstranskripte b2a2 und b3a2 oder die ALL-typische-Variante e1a2. Daneben existiert noch die sehr seltene Bruchpunktvariante µ-bcr. Modifiziert nach 2.
Klinische Bedeutung:
Zur Diagnosesicherung sollte neben der konventionellen Zytogenetik auch eine molekulare Diagnostik durchgeführt werden, die eine deutlich höhere Sensitivität hat und daher für die Erstdiagnose, vor allem aber für die Verlaufskontrolle unter und nach Chemotherapie hilfreich ist. Patienten mit Ph-negativer, BCR-ABL-positiver CML unterscheiden sich weder klinisch noch hämatologisch von Patienten mit klassischer Philadelphia - Translokation. Eine Ph-negative, BCR-ABL-negative CML gilt als prognostisch ungünstig. Bei ALL gilt der Nachweis von BCR-ABL als prognostisch ungünstig.
Monitoring unter Therapie mit Imatinib:
Der BCR-ABL Nachweis zählt zu den Indikationen des Tyrosinkinaseinhibitors Imatinib (Glivec). Unter Therapie mit Imatinib fallen in der Regel die residualen Leukämiezellen in Knochenmarkaspiraten unter die zytogenetische Nachweisgrenze. Mittels quantitativer PCR kann das Ausmaß des molekularen Ansprechens und damit eine Remission bzw. ein Rezidiv frühzeitig erkannt werden. Zusätzliche Chromosomenanomalien im Rahmen der klonalen Evolution der CML können mittels PCR nicht nachgewiesen werden. Diese können durch vollständige Chromosomenanalyse (10 ml unzentrifugiertes Li-Heparinblut) oder, bei spezifischen Fragestellungen, mittels FISH (Fluoreszenz in situ Hybridisierung) aufgedeckt werden.
Material:
Erstdiagnosen: Methode: RT-PCR. Der Test detektiert alle Fusionstransskripte (M-bcr, m-bcr und µ-bcr). EDTA-Blut: 2 ml, EDTA-Knochenmark: 2 ml Verlaufskontrollen: Methode: quantitative RT-PCR EDTA-Blut: 2 ml, EDTA-Knochenmark: 2 ml Wir bitten um deutliche Kennzeichnung, ob es sich um eine Erstdiagnose oder Verlaufskontrolle handelt!
Literatur:
1.) Hochhaus A. et al., Dtsch Med Wochenschr, 2004, 129: 2122
2.) Nashed A. et al., J M D, 2003, 5: 63
3.) Hochhaus A., Best Prac Res Clin Haematol 2002, 15: 159
4.) Cortes J. et al., Cancer, 1995, 75: 464
5.) Hughes T. et al., Blood Reviews, 2006, 20: 29
6.) Onida F. et al., Cancer
Freie Leichtketten im Serum - Verbesserte Diagnostik und Verlaufskontrolle monoklonaler Gammopathien
Hintergrund: Anhand der monoklonalen Gammopathie wird eine Gruppe von Non-Hodgkin-Lymphomen beschrieben, deren Merkmal die Überexpression eines Plasmazellklons ist. Die monoklonalen Immun-globuline (Paraproteine) bestehen aus Schwerketten (α, δ, ε, γ oder μ) sowie den dazu korrespondierenden Leichtketten kappa κ oder lambda λ. Die Leichtketten können gebunden an ihre korrespondierende Schwerkette oder auch frei aus der Plasmazelle ins Serum sezerniert werden. Nach dem Erstbeschreiber werden monoklonal synthetisierte freie Leichtketten im Urin historisch als Bence-Jones-Proteine (BJP) bezeichnet.
Abb. 1: Grundstruktur eines Immunglobulins am Beispiel des IgG: γ Schwerkette, κ oder λ Leichtkette
Diagnostik: Eine monoklonale Gammopathie zeigt sich meist durch das Auftreten eines schmalbasigen Extragradienten in der Serum-Protein-Elektrophorese. Mittels Immunfixationselektrophorese (IFE) im Serum oder Urin können die monoklonalen Paraproteine qualitativ bestätigt werden. Mit der Bestimmung der Gesamt-Leichtketten (frei und gebunden) in Serum und Urin war bisher eine Quantifizierung in hohen Konzentrationsbereichen möglich. Diese Methoden sind allerdings aufgrund ihrer relativ hohen Nachweisgrenzen in ihrer Sensitivität limitiert und können insbesondere bei Patienten mir geringer Ausscheidung von Paraproteinen (MGUS, Residuen, Rezidiv, nicht-sekretorisches MM, Leichtketten MM, AL-Amyloidose) oft falsch negativ sein.
Durch die hochsensitive, quantitative Bestimmung von freien Leichtketten im Serum und Urin verbessert sich die Diagnostik monoklonaler Gammopathien deutlich. Aufgrund der niedrigen Nachweisgrenze von 10 mg/l, der kurzen Halbwertszeit von 2-6 h und der Unabhängigkeit von der Nierenfunktion stellt die Bestimmung der freien Leichtketten eine wichtige Ergänzung sowohl bei der Diagnostik als auch bei Verlaufs- und Therapiekontrolle von monoklonalen Gammopathien dar und sollte daher bei folgenden Indikationen stets in Kombination mit der Elektrophorese/IFE durchgeführt werden:
- Monoklonale Gammopathie mit unbestimmter Signifikanz (MGUS)
Eine Krankheitsprogression kann durch die Bestimmung der freien Leichketten rechtzeitig erkannt werden. - Multiples Myelom (MM):
Bei ca. 95% der MM Patienten sind freie Leichtketten im Serum nachweisbar. - Leichtkettenmyelom (LCCM)
Leichkettenmyelome produzieren ausschließlich freie Leichtketten und sind in der Elektrophorese negativ. Der Nachweis der freien Leichketten in der IFE im Urin ist erst möglich, wenn die Resorptionskapazität im proximalen Nierentubulus überschritten ist - Nicht-sekretorisches MM:
Nicht-sekretorische MM zeigen eine unauffällige IFE, sezernieren aber in 50-70% der Fälle geringe Mengen an freien Leichtketten. - AL-Amyloidose:
Die Amyloidfibrillen bestehen aus monoklonalen Leichtketten, die durch die Bestimmung der freien Leichtketten detektiert werden.
Empfohlene Labordiagnostik:
Diagnose: freie Leichtketten im Serum, ggf. Urin, Elektrophorese, Immunfixationselektrophorese (Serum, ggf. Urin)
Verlaufs-/Therapiekontrolle: freie Leichtketten, Elektrophorese, IFE, Blutbild, LDH, Ca, Immunglobuline quantitativ, Beta-2-Mikroglobulin im Serum, Thymidinkinase, ggf. Proteinurie-differenzierung, ggf. Beta-Cross-Laps i.Serum, Knochenmarkzytologie
Material:
2 ml Serum, ggf. 2 ml Urin
S100: Tumormarker für das maligne Melanom - Marker für zerebrale Schädigung
Hintergrund
Trotz aller Früherkennungsprogramme versterben immer noch ca. 20 % der Melanompatienten an der Krankheitspro-gression. Daher sind Laboruntersuchungen zum Ausschluss von Metastasen, die den Patienten nur wenig belasten, von besonderem Interesse. S100 gehört zu einer multigen-etischen Familie Kalzium-bindender, regulat-orischer Proteine. S100A1 und S100B werden von Melanomzellen sowie Zellen des ZNS und in geringerem Ausmaß auch von anderem Gewebe exprimiert.
Indikation
- malignes Melanom: zur Prognoseeinschätzung, Therapiekon-trolle und Nachsorge (serielle Messungen, s.u.).
- Differenzialdiagnostische Unterstützung bei CUP (cancer of unknown primary origin).
- Hilfe beim Management von Patienten nach potentieller Hirnschädigung zusammen mit weiteren klinischen Informationen und bildgebenden Verfahren.
Bewertung
Die S100 Freisetzung ins Serum beim malignenen Melanom ist stadienabhängig und spiegelt die Tumormasse wieder. Patienten in fortgeschrittenen Stadien weisen höhere Kon-zentrationen auf. Ansteigende S100-Konzentrationen können eine Krankheitspro-gression ca. 5-23 Wochen vor klinischen oder radiologischen Methoden detektieren. Insbesondere bei Patienten im Stadium III weisen ansteigende S100-Serumkon-zentrationen auf Fernmetastasen hin. Eine erfolgreiche Therapie führt zu einem signifikanten Rückgang der S100-Kon-zentrationen. Patienten mit normalen S100-Werten weisen ein signifikant längeres Überlebensintervall auf, als Patienten mit erhöhten Werten.
Daneben kann nach zerebraler Schädigung mit Störung der Blut-Hirn-Schranke die S100-Konzentration im Serum ansteigen.
Untersuchungsintervalle bei Melanomen:
Im Stadium I und II ist eine präoperative Bestimmung indiziert. Die Intervalle der Nachsorge können Sie der Tabelle entnehmen.
Material:
Serum
Labornachsorge kutaner maligner Melanome (AWMF-Leitlinie):
| Stadium | Labor S100B |
| Jahr | 1-3 | 1-3 | 4 + 5 | 6-10 |
| IA | 6-mtl. | - | - | - |
| IB-IIB | 3-mtl. | 3-mtl. | - | - |
| IIC-IV* | 3-mtl. | 3-mtl. | 6-mtl. | - |
Literatur:
1.) Thomas L.: Labor und Diagnose. 2005, TH-Books
2.) Harpio R. et al.: S100 proteins as cancer biomarkers with focus on S100B in malignant melanoma. 2004, Clin Biochem, 37: 512-518
3.) AWMF-Leitlinien, Interdisziplinäre Leitlinien der Deutschen Krebsgesellschaft und der Deutschen Dermatologischen Gesellschaft 02/2005